Regolamento di attuazione delle direttive 96/51/CE, 98/51/CE e 1999/20/CE in materia di additivi nell'alimentazione degli animali
| Settore: | Normativa nazionale |
| Materia: | 4. Alimenti e bevande |
| Capitolo: | 4.4 alimenti per animali |
| Data: | 02/11/2001 |
| Numero: | 433 |
| Sommario |
| Art. 1. Ambito di applicazione |
| Art. 2. Definizioni |
| Art. 3. Condizioni per l'autorizzazione |
| Art. 4. Procedura per l'autorizzazione |
| Art. 5. Valutazione specifica per particolari additivi |
| Art. 6. Autorizzazione comunitaria associata al responsabile |
| Art. 7. Protezione dei dati |
| Art. 8. Autorizzazione comunitaria non associata al responsabile |
| Art. 9. Additivi già autorizzati |
| Art. 10. Tenori degli additivi e loro miscele |
| Art. 11. Sperimentazioni a fini scientifici |
| Art. 12. Norme per la conservazione di additivi e premiscele |
| Art. 13. Provvedimenti di sospensione |
| Art. 14. Tenori di taluni additivi nei mangimi complementari |
| Art. 15. Immissione in commercio |
| Art. 16. Etichettatura degli additivi |
| Art. 17. Etichettatura delle premiscele |
| Art. 18. Etichettatura dei mangimi |
| Art. 19. Ulteriori norme sui mangimi complementari |
| Art. 20. Vigilanza e controlli |
| Art. 21. Norme transitorie |
| Art. 22. Disposizioni finanziarie |
| Art. 23. Abrogazioni |
§ 4.4.26 - D.P.R. 2 novembre 2001, n. 433. [1]
Regolamento di attuazione delle direttive 96/51/CE, 98/51/CE e 1999/20/CE in materia di additivi nell'alimentazione degli animali
(G.U. 15 dicembre 2001, n. 291, S.O.)
Art. 1. Ambito di applicazione
1. Il presente regolamento disciplina la preparazione, il commercio, la distribuzione, anche a titolo gratuito, e l'impiego degli additivi, delle premiscele e dei mangimi che li contengono, utilizzabili nell'alimentazione degli animali.
2. Il presente regolamento non si applica ai coadiuvanti tecnologici utilizzati deliberatamente come sostanze nella trasformazione di materie prime per mangimi o di mangimi ai fini di un determinato obiettivo tecnologico, durante il trattamento o la trasformazione, e il cui impiego può risultare nella presenza non intenzionale, ma tecnicamente inevitabile, di residui di tali sostanze o di loro derivati nel prodotto finale, purché i suddetti residui non presentino rischi sanitari e non abbiano alcun effetto tecnologico sul prodotto finito.
3. Non sono considerati additivi le sostanze che, pur corrispondendo ad una sostanza autorizzata ai sensi del presente regolamento, sono presenti allo stato naturale nella materia prima, che rientrano nella composizione normale dei mangimi, purché non si tratti di prodotti specificamente arricchiti con tali sostanze.
4. Il presente regolamento non si applica agli additivi, alle premiscele ed ai mangimi destinati all'esportazione; tale destinazione deve essere dimostrata con una indicazione appropriata, anche sull'imballaggio, sul recipiente e sui documenti di accompagnamento.
Art. 2. Definizioni
1. Ai fini del presente regolamento si intende per:
a) "additivi": le sostanze o le preparazioni utilizzate nell'alimentazione degli animali che hanno una o più delle seguenti finalità di seguito elencate:
1) influenzare favorevolmente le caratteristiche delle materie prime per mangimi o dei mangimi composti o dei prodotti di origine animale;
2) soddisfare le esigenze nutrizionali degli animali o migliorare la produzione animale influendo, in particolare, sulla flora gastrointestinale o sulla digeribilità dei mangimi;
3) introdurre elementi favorevoli per raggiungere obiettivi nutrizionali particolari o per rispondere a esigenze nutrizionali specifiche momentanee degli animali;
4) prevenire o ridurre gli effetti nocivi provocati dalle deiezioni animali oppure migliorare l'ambiente in cui si trovano gli animali;
b) "materie prime per mangimi": i diversi prodotti di origine vegetale o animale, allo stato naturale, freschi o conservati nonché i derivati della loro trasformazione industriale, come pure le sostanze organiche o inorganiche, comprendenti o no additivi destinati ad essere impiegati nell'alimentazione degli animali per via orale, direttamente come tali o previa trasformazione, per la preparazione di mangimi composti oppure ad essere usati come supporto delle premiscele;
c) "premiscele": le miscele di additivi o le miscele di uno o più additivi con sostanze che costituiscono un supporto, destinate alla fabbricazione di mangimi. Il termine "premiscela" sostituisce il termine "integratore" utilizzato nella
d) "immissione in circolazione" ovvero "circolazione": la detenzione di prodotti a fini di vendita, ivi compresa l'offerta, o altre forme di trasferimento a terzi, a titolo gratuito o a titolo oneroso, nonché la vendita e le altre forme di trasferimento;
e) "responsabile dell'immissione in circolazione": la persona fisica o giuridica responsabile della conformità dell'additivo ai requisiti oggetto di autorizzazione comunitaria e della relativa immissione in circolazione;
f) "microrganismi": i microrganismi che formano colonie;
g) "additivi oggetto di autorizzazione associata al responsabile della loro immissione in circolazione": gli additivi di cui all'allegato C, parte I
h) "altri additivi": gli additivi che non sono oggetto di un'autorizzazione associata al responsabile della loro immissione in circolazione e di cui all'allegato C, parte II.
2. Per le definizioni di mangimi, mangimi composti, mangimi completi, mangimi complementari, razioni giornaliere, animali e animali familiari valgono le definizioni di cui all'allegato I alla
Art. 3. Condizioni per l'autorizzazione
1. Nessun additivo può essere immesso in circolazione senza un'apposita autorizzazione comunitaria, rilasciata con regolamento della Commissione europea a seguito della procedura di cui all'articolo 4.
2. L'autorizzazione comunitaria di cui al comma 1 è rilasciata a condizione che:
a) l'additivo utilizzato nei mangimi abbia uno degli effetti di cui all'articolo 2, comma 1, lettera a);
b) l'additivo, tenuto conto delle condizioni di impiego, non abbia influenze sfavorevoli sulla salute umana o animale o sull'ambiente e non danneggi il consumatore alterando le caratteristiche dei prodotti di origine animale;
c) l'additivo sia controllabile sia in quanto additivo stesso, sia nelle premiscele, sia nei mangimi o, sia, eventualmente, nelle materie prime per mangimi;
d) l'additivo, tenuto conto del tenore consentito, non possa essere usato per il trattamento o la prevenzione delle malattie degli animali; questa condizione non si applica agli additivi appartenenti al gruppo dei coccidiostatici e altre sostanze medicamentose;
e) per seri motivi attinenti alla salute umana o degli animali, l'additivo non sia esclusivamente riservato all'uso medico o veterinario.
3. Gli additivi autorizzati in base alle disposizioni del presente decreto possono essere immessi in circolazione e possono essere utilizzati alle condizioni previste nel relativo regolamento di autorizzazione solo se incorporati nei mangimi.
4. In deroga al comma 3, gli additivi appartenenti a gruppi diversi da "antibiotici, "coccidiostatici e altre sostanze medicamentose", nonché "fattori di crescita", possono essere utilizzati secondo modi di somministrazione diversi dall'incorporazione nei mangimi, purché questi siano previsti dal regolamento di autorizzazione.
5. Gli additivi non devono essere aggiunti alle materie prime per mangimi e ai mangimi semplici a meno che la loro utilizzazione non sia espressamente prevista nel regolamento di autorizzazione.
6. L'autorizzazione degli additivi di cui all'articolo 2, comma 1, lettera g), comporta l'attribuzione di uno o più numeri di immatricolazione per ciascun responsabile dell'immissione in circolazione e di un numero di registrazione CE dell'additivo.
7. L'autorizzazione degli additivi di cui all'articolo 2, comma 1, lettera h), comporta l'attribuzione di un numero di registrazione CE dell'additivo.
8. L'autorizzazione dell'additivo è revocata mediante regolamento della Commissione europea:
a) su richiesta del responsabile dell'immissione in circolazione dell'additivo, se si tratta di un additivo di cui all'articolo 2, comma 1, lettera g);
b) se non è più soddisfatta una delle condizioni connesse all'autorizzazione dell'additivo di cui al comma 2;
c) se un campione standard dell'additivo non è fornito alle autorità competenti degli Stati membri che lo hanno richiesto o se l'additivo immesso in circolazione non corrisponde al campione standard dell'additivo autorizzato;
d) se un campione di riferimento della sostanza attiva non è fornito alle autorità competenti degli Stati membri che lo hanno richiesto;
e) se il responsabile dell'immissione in circolazione dell'additivo non fornisce, entro un termine determinato, le informazioni richieste dalla Commissione europea.
9. La Commissione europea può consentire, per un anno al massimo, lo smaltimento delle scorte dell'additivo la cui autorizzazione è stata revocata ai sensi del comma 8, se continuano ad essere soddisfatte almeno le condizioni di cui al comma 2, lettere b) ed e).
Art. 4. Procedura per l'autorizzazione
1. Chi intende ottenere l'autorizzazione comunitaria di cui all'articolo 3 di una sostanza o di una preparazione quale additivo o, nel caso di un additivo già utilizzato di una nuova autorizzazione, sceglie uno Stato membro quale relatore, in occasione della procedura d'esame, del fascicolo da lui costituito conformemente alle disposizioni dell'allegato D. Per l'Italia l'autorità competente è il Ministero della salute; il richiedente che opera in un Paese terzo deve disporre di un rappresentante stabilito nella Comunità europea.
2. Il Ministero della salute, qualora incaricato come relatore ai sensi del comma 1, verifica che il fascicolo sia costituito in base alle disposizioni dell'allegato D e che la sostanza o la preparazione, stando ai dati forniti, sia conforme alle condizioni previste all'articolo 3, comma 2.
3. Il richiedente, per il tramite del Ministero della salute, entro un anno dalla data di notifica del fascicolo di cui al comma 1, fatto salvo rigetto o rinvio, trasmette la domanda corredata del fascicolo alla Commissione europea e agli altri Stati membri ai fini dell'emanazione del regolamento di cui all'articolo 3, comma 1; questi ultimi, entro sessanta giorni, verificano la conformità del fascicolo all'allegato D e trasmettono per iscritto eventuali osservazioni alla Commissione europea e agli altri Stati membri; tale termine si applica anche al Ministero della salute, nel caso in cui non sia stato scelto quale relatore del fascicolo.
4. Il Ministero della salute informa il richiedente, la Commissione europea e gli altri Stati membri dei motivi dell'eventuale rigetto o rinvio del fascicolo.
5. Le disposizioni di cui alla
6. Le disposizioni di cui al comma 5, non si applicano:
a) alle denominazioni e alla composizione dell'additivo;
b) alle proprietà fisico-chimiche e biologiche dell'additivo;
c) all'interpretazione dei dati farmacologici, tossicologici ed ecotossicologici dell'additivo;
d) ai metodi di analisi per il controllo dell'additivo stesso, dell'additivo nelle premiscele, nei mangimi ed, eventualmente, nelle materie prime;
e) ai metodi di controllo dei residui dell'additivo o di suoi metaboliti nei prodotti di origine animale.
7. Per gli additivi di cui all'articolo 2, comma 1, lettera g), il richiedente l'autorizzazione di cui al comma 1 deve presentare una monografia conforme all'allegato D.
8. La monografia di cui al comma 7 è utilizzata per accertare se l'additivo del quale si chiede l'autorizzazione all'immissione in circolazione costituisce un prodotto nuovo o deve essere considerato una copia o per verificare se l'additivo immesso in circolazione corrisponde all'additivo descritto nel fascicolo in base al quale l'autorizzazione comunitaria è stata rilasciata.
9. Alla richiesta di autorizzazione all'immissione in circolazione di cui al comma 1 deve essere allegata una scheda che riassume le caratteristiche e le proprietà dell'additivo, conformemente alle disposizioni di cui all'allegato D; per gli additivi di cui all'articolo 2, comma 1, lettera g), la scheda deve contenere le caratteristiche e le proprietà più importanti indicate nella monografia.
10. Le schede di cui al comma 9 sono pubblicate nella Gazzetta Ufficiale delle Comunità europee.
11. Per gli additivi di cui all'articolo 2, comma 1, lettera g), il responsabile dell'immissione in circolazione deve mettere a disposizione dell'Istituto superiore di sanità, su richiesta del Ministero della salute, un campione standard avente le caratteristiche e le proprietà dell'additivo descritte nella monografia di cui al comma 7 e un campione di riferimento della sostanza attiva. In caso di modifica delle caratteristiche o delle proprietà dell'additivo, deve essere depositato un nuovo campione standard corrispondente alla nuova monografia dell'additivo.
Art. 5. Valutazione specifica per particolari additivi
1. Qualora un additivo sia costituito da o contenga organismi geneticamente modificati, di cui all'articolo 1 del
a) copia di ogni provvedimento formale di assenso del Ministero della salute per l'emissione deliberata nell'ambiente degli organismi geneticamente modificati per scopi di ricerca e sviluppo, conformemente all'articolo 6, comma 4, del
b) un fascicolo tecnico completo che fornisca le informazioni previste negli allegati II e III del
2. Gli articoli da 11 a 17 del
Art. 6. Autorizzazione comunitaria associata al responsabile
1. Gli additivi di cui all'articolo 2, comma 1, lettera g), che soddisfano le condizioni previste dall'articolo 3, comma 2, sono autorizzati e iscritti nel capitolo I "Elenco degli additivi associati al responsabile dell'immissione in circolazione cui è concessa una autorizzazione per 10 anni" di cui al comma 9.
2. Per gli additivi di cui all'articolo 2, comma 1, lettera g), l'autorizzazione comunitaria può essere concessa in via provvisoria per una durata non superiore ai quattro anni, purché siano soddisfatte le condizioni previste all'articolo 3, comma 2, lettere b), c), d) ed e), e si possa presumere, in base ai risultati disponibili, che la condizione indicata alla lettera a) dello stesso articolo sia anch'essa soddisfatta.
3. L'autorizzazione in via provvisoria degli additivi immessi in circolazione anteriormente alla data del 1° aprile 1998, ha la durata massima di cinque anni.
4. Gli additivi autorizzati ai sensi dei commi 2 e 3 sono iscritti nel capitolo II "Elenco degli additivi associati al responsabile dell'immissione in circolazione cui è concessa un'autorizzazione a titolo provvisorio" di cui al comma 9.
5. L'autorizzazione comunitaria per gli additivi di cui all'articolo 2, comma 1, lettera g), è valida per dieci anni a decorrere dalla data in cui ha effetto l'autorizzazione definitiva e può essere rinnovata di decennio in decennio. In caso di rinnovo, il titolare dell'autorizzazione presenta alla Commissione europea, e in copia agli altri Stati membri, almeno un anno prima della data di scadenza dell'autorizzazione e per il tramite del Ministero della salute, una domanda corredata di un fascicolo conforme a quanto stabilito in sede comunitaria.
6. Alle domande di rinnovo si applicano le disposizioni di cui agli articoli 3, 4, 5 e 22.
7. Qualora, per motivi non attribuibili al titolare dell'autorizzazione, non si possa decidere sulla domanda di rinnovo prima della data di scadenza dell'autorizzazione, la durata di quest'ultima è prorogata automaticamente sino al momento delle determinazioni della Commissione europea.
8. Il responsabile dell'immissione in circolazione degli additivi di cui all'articolo 2, comma 1, lettera g), trasmette alla Commissione europea i nomi o le ragioni sociali e l'indirizzo o la sede sociale dei soggetti cui essi hanno demandato la fabbricazione degli additivi nonché, se tali soggetti sono stabiliti in un Paese terzo, anche il nome o la ragione sociale nonché l'indirizzo o la sede sociale dei loro rappresentanti nella Comunità europea.
9. L'elenco dei soggetti di cui al comma 8 nonché l'elenco degli additivi autorizzati sono pubblicati nella Gazzetta Ufficiale delle Comunità europee.
Art. 7. Protezione dei dati
1. I dati scientifici e le informazioni sugli additivi di cui all'articolo 2, comma 1, lettera g), contenuti nel fascicolo presentato ai fini del rilascio della prima autorizzazione, fatti salvi eventuali accordi tra gli interessati, non possono essere utilizzati da altri richiedenti per un periodo di dieci anni a decorrere:
a) per gli additivi di cui all'articolo 9, dalla data della prima autorizzazione concessa con regolamento della Commissione europea;
b) per gli altri additivi, dalla data della prima autorizzazione concessa con regolamento della Commissione europea o a decorrere dal 1° ottobre 1999, se tale data è precedente.
2. In deroga al comma 1, prima della scadenza dei dieci anni, possono essere rilasciate ad altri richiedenti autorizzazioni all'immissione in circolazione relative allo stesso additivo, purché siano soddisfatte le condizioni dell'articolo 3, comma 2, e dell'articolo 4.
3. I dati complementari di un additivo oggetto di autorizzazione provvisoria sono considerati parte integrante del fascicolo iniziale e i termini di scadenza della relativa protezione coincidono con quelli contenuti nel fascicolo iniziale.
4. Allo scadere del periodo di cui al comma 1, i risultati complessivi o parziali della valutazione effettuata in base ai dati scientifici e alle informazioni contenuti nel fascicolo presentato ai fini del rilascio dell'autorizzazione di cui all'articolo 3, comma 1, possono essere utilizzati dalla Commissione europea o da uno Stato membro a vantaggio di un altro richiedente l'autorizzazione per l'immissione in circolazione di un additivo già autorizzato.
5. Nell'ipotesi di cui al comma 4, conformemente alle disposizioni adottate in sede comunitaria, il nuovo richiedente trasmette, all'autorità competente di cui all'articolo 4, la domanda e il relativo fascicolo.
6. Le disposizioni di cui ai commi 4 e 5 si applicano anche in caso di utilizzazione dei dati di un fascicolo relativi ad un additivo oggetto di una revoca di autorizzazione, su richiesta del titolare di tale autorizzazione.
7. I dati scientifici e le informazioni supplementari presentati al fine di modificare le condizioni di iscrizione dell'additivo per il rinnovo dell'autorizzazione ai sensi dell'articolo 6, comma 5, o qualsiasi dato scientifico nuovo o informazioni fornite durante il periodo di autorizzazione dell'additivo non devono essere utilizzati dalla Commissione europea o da uno Stato membro a beneficio di un altro richiedente per un periodo non superiore ai cinque anni a decorrere dalla data dalla quale ha effetto l'autorizzazione di una nuova utilizzazione, il rinnovo o il deposito dei nuovi dati scientifici o informazioni.
8. Se il periodo di protezione dei dati di cui al comma 7, concesso per una modifica delle condizioni di iscrizione di un additivo, scade prima di quello di cui al comma 1, il periodo di cinque anni è prorogato in modo da far coincidere la scadenza dei due termini.
9. Fatte salve le disposizioni di cui al comma 1, il richiedente l'autorizzazione per un additivo di cui all'articolo 2, comma 1, lettera g), deve verificare, prima di avviare esperimenti tossicologici su animali vertebrati, se il prodotto o la sostanza attiva non sia già stata autorizzata; a tal fine, può informarsi presso l'autorità competente degli Stati membri.
10. Qualora si tratti di un prodotto o di una sostanza attiva già autorizzata, il richiedente e i titolari di precedenti autorizzazioni si adoperano per pervenire ad un accordo sull'impiego in comune delle informazioni, in modo da evitare la ripetizione degli esperimenti tossicologici sugli animali vertebrati.
Art. 8. Autorizzazione comunitaria non associata al responsabile
1. Gli additivi di cui all'articolo 2, comma 1, lettera h), che soddisfano le condizioni di cui all'articolo 3, comma 2, sono autorizzati e iscritti nel capitolo III dell'"Elenco degli altri additivi per i quali l'autorizzazione è concessa senza limiti di tempo" di cui all'articolo 6, comma 9.
2. Gli additivi di cui all'articolo 2, comma 1, lettera h), già iscritti nell'allegato I del decreto del Presidente della Repubblica 1° marzo 1992, n. 228, e successive modificazioni, anteriormente al 1° aprile 1998, sono autorizzati e iscritti nel capitolo III dell'elenco di cui al comma 1.
3. Per gli additivi di cui all'articolo 2, comma 1, lettera h), l'autorizzazione comunitaria può essere concessa in via provvisoria per una durata non superiore ai quattro anni, purché siano soddisfatte le condizioni previste all'articolo 3, comma 2, lettere b), c), d), ed e), e si possa presumere che la condizione indicata alla lettera a) dello stesso articolo è soddisfatta; tali additivi sono iscritti nel capitolo IV "Elenco degli altri additivi per i quali l'autorizzazione è concessa a titolo provvisorio", di cui all'articolo 6, comma 9.
4. L'autorizzazione provvisoria nazionale degli additivi di cui all'articolo 2, comma 1, lettera h), già iscritti nell'allegato II del citato decreto n. 228 del 1992 anteriormente al 1° aprile 1998, continua ad essere valida; tali additivi sono iscritti nel capitolo IV dell'elenco di cui al comma 3. La durata dell'autorizzazione provvisoria di tali additivi non può essere superiore a cinque anni tenuto conto del periodo di iscrizione nel succitato allegato II.
Art. 9. Additivi già autorizzati
1. In deroga all'articolo 3, comma 1, è consentita l'immissione in circolazione degli additivi di cui all'articolo 2, comma 1, lettera g), già iscritti nell'allegato 1 del decreto del Presidente della Repubblica 1° marzo 1992, n. 228, anteriormente al 1° gennaio 1988 e indicati nell'allegato B tali additivi sono autorizzati provvisoriamente in attesa che la Commissione europea riesamini le autorizzazioni, su istanza del richiedente, secondo le procedure previste dal presente regolamento.
Art. 10. Tenori degli additivi e loro miscele
1. I tenori massimi e minimi stabiliti per taluni additivi si riferiscono ai mangimi completi con tasso di umidità del 12 per cento, quando i regolamenti comunitari di autorizzazione non prevedono disposizioni particolari.
2. Se la sostanza utilizzabile come additivo esiste anche allo stato naturale in talune materie prime del mangime, la parte di additivo da incorporare deve essere calcolata in modo che la somma degli elementi aggiunti e degli elementi presenti naturalmente non superi il tenore massimo previsto nei regolamenti comunitari di autorizzazione.
3. Nelle premiscele e nei mangimi è ammessa la miscelazione degli additivi unicamente se viene rispettata la compatibilità fisico-chimica e biologica tra i componenti della miscela, in funzione degli effetti ricercati.
4. Se non si tratta di una miscela oggetto di autorizzazione specifica in quanto additivo, non possono essere mescolati tra loro:
a) gli antibiotici e i fattori di crescita, sia che appartengano ad uno stesso gruppo, sia che appartengano ai due gruppi;
b) i coccidiostatici e le altre sostanze medicamentose, con gli antibiotici ed i fattori di crescita, quando gli stessi coccidiostatici esercitano, per una stessa categoria di animali, una funzione di antibiotico o di fattore di crescita;
c) i coccidiostatici e le altre sostanze medicamentose, se i loro effetti sono analoghi.
5. La miscela di antibiotici, fattori di crescita, coccidiostatici e altre sostanze medicamentose con microrganismi è vietata a meno che nel regolamento comunitario di autorizzazione del microrganismo non sia ammessa tale miscela.
Art. 11. Sperimentazioni a fini scientifici
1. In deroga all'articolo 3 e all'articolo 10, commi 3 e 4, il Ministero della salute può autorizzare, esclusivamente per esperimenti ai fini scientifici ed a fini non commerciali, l'utilizzazione come additivi di prodotti non autorizzati a livello comunitario o l'utilizzazione di additivi a condizioni diverse da quelle previste nel regolamento comunitario purché gli esperimenti siano effettuati secondo i principi e le condizioni fissati in sede comunitaria e sotto il controllo delle aziende unità sanitarie locali competenti per territorio, secondo le modalità previste dal
2. Le deroghe di cui al comma 1 sono consentite solo nel caso in cui la sperimentazione di tali prodotti non comporti un rischio per la salute dell'uomo, dell'animale o dell'ambiente.
3. Il Ministero della salute può consentire l'utilizzazione nell'alimentazione umana di prodotti ottenuti da animali oggetto delle sperimentazioni di cui ai commi 1 e 2.
Art. 12. Norme per la conservazione di additivi e premiscele
1. Gli additivi e le premiscele devono essere adeguatamente custoditi e contenuti in recipienti particolarmente idonei alla loro conservazione che possono essere facilmente identificati.
2. Gli additivi e le premiscele di cui al comma 1 devono essere commercializzati in imballaggi o recipienti sigillati il cui dispositivo di chiusura sia tale da non poter essere riutilizzato dopo l'apertura.
Art. 13. Provvedimenti di sospensione
1. L'impiego di uno degli additivi autorizzati o la sua utilizzazione alle condizioni eventualmente fissate possono essere sospesi provvisoriamente o limitati nel territorio quando si constati che essi comportano un pericolo per la salute dell'uomo o degli animali o per l'ambiente.
2. Il Ministro della salute e il Ministro dell'ambiente e della tutela del territorio, con proprie ordinanze e secondo le rispettive competenze, provvedono a quanto previsto al comma 1.
3. L'adozione del provvedimento di cui al comma 2 e i motivi che lo giustificano sono comunicati alla Commissione europea e agli altri Stati membri a cura del Ministero competente.
4. Qualora la Commissione europea avvii la procedura per adottare le necessarie modifiche, le misure adottate nell'ordinanza di cui al comma 2 possono essere mantenute fino all'entrata in vigore delle modifiche stesse.
Art. 14. Tenori di taluni additivi nei mangimi complementari
1. I mangimi complementari, tenuto conto della diluizione prevista per il loro impiego, non possono contenere tenori di additivi superiori a quelle fissati per i mangimi completi.
2. Nei mangimi complementari i tenori di antibiotici, di coccidiostatici ed altre sostanze medicamentose, di fattori di crescita, di vitamina D e di antiossidanti possono superare i tenori massimi fissati per i mangimi completi solo in uno dei seguenti casi:
a) se si tratta di mangimi complementari a disposizione di tutti gli utilizzatori, a condizione che il loro tenore di antibiotico o di vitamina D o di fattore di crescita non superi il quintuplo del tenore massimo fissato;
b) se si tratta di mangimi complementari destinati a talune specie animali a disposizione di tutti gli utilizzatori in considerazione del sistema particolare di nutrizione, a condizione che la percentuale non superi:
1) per gli antibiotici ed i fattori di crescita 1000 mg/kg e per i bovini destinati all'ingrasso, 2000 mg/kg;
2) per gli antiossidanti, nonché per i coccidiostatici ed altre sostanze medicamentose, il quintuplo del tenore massimo fissato;
3) per le vitamine D, 200.000 Ul/kg.
3. Nel caso di cui al comma 2, il mangime deve presentare nella composizione una o più caratteristiche, quali proteine o minerali, che escludano il superamento dei tenori di additivi fissati per i mangimi completi o la destinazione del mangime ad altre specie animali.
4. La distribuzione e l'utilizzo dei mangimi complementari di cui al comma 2, sono consentiti solo previo riconoscimento o registrazione ai sensi del
Art. 15. Immissione in commercio
1. Possono immettere in circolazione o utilizzare i relativi additivi contemplati dal presente regolamento, le premiscele preparate con questi additivi per essere incorporate nei mangimi composti, nonché i mangimi composti contenenti queste premiscele, soltanto le imprese o gli intermediari che soddisfano le condizioni previste dal
2. Gli additivi di cui all'allegato A, parte I, possono essere forniti soltanto da imprese riconosciute ai sensi del
a) ad intermediari o ad imprese di fabbricazione di premiscele che sono stati riconosciuti ai sensi rispettivamente dell'articolo 3, comma 1, e dell'articolo 2, comma 2, lettera b), del
b) sotto forma di premiscele, soltanto ad intermediari o ad imprese che procedono alla fabbricazione di mangimi composti, al fine della loro immissione in circolazione o esclusivamente per le necessità del bestiame ivi allevato, riconosciuti in base alle disposizioni rispettivamente dell'articolo 3, comma 1, e dell'articolo 2, comma 2, lettere c) o e), del citato decreto n. 123 del 1999.
3. Gli additivi di cui all'allegato A, parte II, possono essere forniti soltanto da imprese riconosciute:
a) ad intermediari o ad imprese di fabbricazione di premiscele che sono stati riconosciuti in base alle disposizioni rispettivamente dell'articolo 3, comma 1, e dell'articolo 2, comma 2, lettera b), del citato decreto n. 123 del 1999;
b) sotto forma di premiscele, soltanto ad intermediari riconosciuti a norma dell'articolo 3, comma 1, del citato
4. Gli additivi di cui all'allegato A, parti I e II, possono essere incorporati nei mangimi composti soltanto se sono stati preventivamente preparati sotto forma di premiscele, in stabilimenti che soddisfano le condizioni previste dall'articolo 2, comma 2, lettera b), del
5. In deroga al comma 4, il Ministero della salute può consentire che siano incorporate alcune premiscele nei mangimi composti in proporzione minore allo 0,2 per cento in peso, ma non al di sotto dello 0,05 per cento in peso, a condizione che la composizione quantitativa e qualitativa delle premiscele lo consenta e qualora sia stato preventivamente accertato dalla regione o dalla provincia autonoma, ai sensi del
6. In deroga ai commi 2 e 3 è consentito fornire:
a) gli additivi di cui all'allegato A, parte II, ad intermediari riconosciuti o a stabilimenti registrati per la fabbricazione di mangimi composti per animali da compagnia che soddisfino le condizioni previste, secondo i casi, dell'articolo 3, comma 1, o dall'articolo 7, comma 2, lettere c) o d), del
b) gli additivi cui all'allegato A, parte I o II, agli stabilimenti per la fabbricazione di mangimi composti, alle seguenti condizioni:
1) che il regolamento di autorizzazione comunitaria dell'additivo ne preveda, per una preparazione specifica dello stesso, l'aggiunta diretta nei mangimi composti;
2) che il fabbricante di mangimi composti sia riconosciuto ai sensi dell'articolo 2, comma 2, lettera c), del
3) che sia stato accertato, previo sopralluogo da parte della regione o provincia autonoma, ai sensi del
7. L'iscrizione degli stabilimenti di cui al comma 6, lettera b), nell'allegato II al
8. La vigilanza e le ispezioni finalizzate al controllo dell'applicazione del presente articolo sono effettuate secondo quanto disposto dal
9. Nelle more della predisposizione degli appositi elenchi comunitari, i prodotti di cui all'articolo 2, comma 2, lettere a), b), c) e d), ed all'articolo 7, comma 2, lettere a), b) e c), del
a) possiede requisiti equivalenti a quelli previsti, secondo il prodotto in questione, all'articolo 2, comma 2, lettere a), b), c) e d), ed all'articolo 7, comma 2, lettere a), b) e c), del
b) dispone di un rappresentante stabilito in Italia, il cui nome e indirizzo deve essere indicato a fronte del nome e indirizzo di ciascuno stabilimento.
10. Il rappresentante di cui al comma 9, lettera b), deve tenere una registrazione dei prodotti che immette in commercio per conto dello stabilimento rappresentato, conforme alle disposizioni contenute negli allegati al
11. Le competenti autorità curano la diffusione dell'elenco di cui al comma 9, senza oneri aggiuntivi per la finanza pubblica.
12. Gli stabilimenti di fabbricazione siti in Paesi terzi che esportano in Italia i prodotti di cui al comma 9 in base alla previgente normativa devono essere resi conformi alle disposizioni di cui al comma 9 entro sei mesi dalla data di entrata in vigore del presente decreto.
Art. 16. Etichettatura degli additivi
1. Gli additivi autorizzati possono essere commercializzati per essere impiegati nell'alimentazione degli animali soltanto se figurano, sull'imballaggio, sul recipiente o su una etichetta ivi fissata in modo ben visibile, chiaramente leggibile ed indelebile, le seguenti indicazioni:
a) per tutti gli additivi, ad eccezione degli enzimi e dei microrganismi:
1) il nome specifico attribuito all'additivo all'atto dell'autorizzazione, il numero di registrazione CE dell'additivo e, nel caso di additivi di cui all'articolo 2, comma 1, lettera g), la denominazione commerciale e il numero di immatricolazione del responsabile dell'immissione in circolazione;
2) il nome o la ragione sociale e l'indirizzo o la sede sociale del responsabile delle indicazioni di cui al presente comma;
3) il peso netto e, per gli additivi liquidi il volume netto, oppure il peso netto;
4) a seconda dei casi, il numero di riconoscimento CE o il numero di registrazione attribuiti allo stabilimento o all'intermediario, ai sensi rispettivamente degli articoli 4 e 9 del
b) oltre a quanto previsto alla lettera a):
1) per gli antibiotici, fattori di crescita, i coccidiostatici e le altre sostanze medicamentose: il nome o la ragione sociale e l'indirizzo o la sede sociale del fabbricante, se quest'ultimo non è responsabile delle indicazioni di etichettatura, il tenore di sostanza attiva, la data limite di garanzia o la durata di conservazione a decorrere dalla data di fabbricazione, il numero di riferimento e la data di fabbricazione della partita, le istruzioni per l'uso ed eventualmente una raccomandazione relativa alla sicurezza di impiego quando gli additivi sono oggetto di disposizioni autorizzatorie particolari;
2) per la vitamina E: il tenore in alfa-tocoferolo e la data limite di garanzia del tenore o la durata di conservazione a decorrere dalla data di fabbricazione;
3) per le vitamine diverse dalla vitamina E, le provitamine e le sostanze aventi un effetto chimico analogo: il tenore di sostanza attiva e la data limite di garanzia del tenore o la durata di conservazione a decorrere dalla data di fabbricazione;
4) per gli oligoelementi, i coloranti compresi i pigmenti, i conservanti e gli altri additivi ad eccezione di quelli appartenenti ai gruppi degli enzimi e dei microrganismi: il tenore di sostanze attive;
c) per gli additivi appartenenti ai gruppi:
1) degli enzimi: il nome specifico del o dei componenti attivi secondo la sua o le loro attività enzimatiche, in base all'autorizzazione concessa, il numero di identificazione secondo l'International Union of Biochemistry, le unità di attività espresse in micromoli di prodotto liberato al minuto per grammo o per millilitro di prodotto enzimatico, il numero di registrazione CE dell'additivo, il nome e la ragione sociale e l'indirizzo o la sede sociale del responsabile delle indicazioni di etichettatura, il nome o la ragione sociale e l'indirizzo o la sede sociale del fabbricante, se quest'ultimo non è responsabile delle indicazioni di etichettatura, il numero di riconoscimento attribuito allo stabilimento o all'intermediario, ai sensi dell'articolo 4 del citato
2) dei microrganismi: l'identificazione del o dei ceppi in base all'autorizzazione concessa, il numero di deposito del o dei ceppi, il numero di unità che formano colonie (CFU/g), il numero di registrazione CE dell'additivo, il nome o la ragione sociale e l'indirizzo o la sede sociale del responsabile dell'etichettatura, il nome o la ragione sociale e l'indirizzo o la sede sociale del fabbricante se quest'ultimo non è responsabile delle indicazioni di etichettatura, il numero di riconoscimento attribuito allo stabilimento o all'intermediario, ai sensi dell'articolo 4 del citato
2. Fatto salvo quanto previsto dal comma 1, la denominazione specifica degli additivi può essere accompagnata:
a) dalla denominazione commerciale;
b) dal nome o dalla ragione sociale e dall'indirizzo o dalla sede sociale del fabbricante se quest'ultimo non è responsabile delle indicazioni di etichettatura, dalle istruzioni per l'uso e dalle eventuali raccomandazioni concernenti la sicurezza d'impiego.
3. Gli imballaggi, i recipienti e le etichette possono contenere indicazioni diverse da quelle prescritte o ammesse a norma dei commi 1 e 2, purché esse siano nettamente separate dalle indicazioni di cui sopra.
4. Il produttore, il condizionatore, l'importatore, il venditore o il distributore stabiliti nel territorio comunitario sono responsabili dell'applicazione delle disposizioni del presente articolo.
5. Per la commercializzazione fra gli Stati membri le indicazioni di cui al presente articolo saranno redatte almeno in una delle lingue ufficiali del Paese destinatario; per la commercializzazione in Italia, tali indicazioni devono essere redatte in lingua italiana.
Art. 17. Etichettatura delle premiscele
1. Le premiscele possono essere commercializzate soltanto se figurano, sull'imballaggio, sul recipiente o sull'etichetta ivi fissata in modo ben visibile, chiaramente leggibile ed indelebile, le seguenti indicazioni:
a) per tutte le premiscele:
1) la denominazione "premiscela";
2) le istruzioni per l'uso e eventualmente una raccomandazione concernente la sicurezza d'impiego delle premiscele;
3) la specie animale o la categoria di animali cui è destinata la premiscela;
4) il nome e la ragione sociale e l'indirizzo o la sede sociale del responsabile delle indicazioni di cui al presente articolo;
5) il peso netto e, per gli additivi liquidi il volume netto, oppure il peso netto;
6) a seconda dei casi, il numero di riconoscimento attribuito all'impresa o all'intermediario, ai sensi dell'articolo 4 del citato
b) oltre a quanto previsto alla lettera a), per le premiscele in cui sono stati incorporati gli additivi sottoindicati:
1) antibiotici, fattori di crescita, coccidiostatici e altre sostanze medicamentose: il nome o la ragione sociale e l'indirizzo o la sede sociale del fabbricante, se costui non è responsabile delle indicazioni di etichettatura, il nome specifico attribuito all'additivo all'atto dell'autorizzazione, il tenore di sostanze attive, la data limite di garanzia del tenore o la durata di conservazione a decorrere dalla data di fabbricazione;
2) sostanze che hanno effetti antiossidanti: il nome specifico attribuito all'additivo all'atto dell'autorizzazione e il tenore di sostanze attive, purché all'atto dell'autorizzazione dell'additivo sia fissato un tenore massimo per i mangimi completi;
3) coloranti, compresi i pigmenti: il nome specifico attribuito all'additivo all'atto dell'autorizzazione e il tenore di sostanze attive, purché all'atto dell'autorizzazione dell'additivo sia fissato un tenore massimo per i mangimi completi;
4) vitamina E: il nome specifico dell'additivo all'atto dell'autorizzazione e il tenore di alfa-tocoferoli e la data limite di garanzia del tenore o la durata di conservazione a decorrere dalla data di fabbricazione;
5) vitamine diverse dalla vitamina E, provitamine e sostanze aventi un effetto analogo: il nome specifico dell'additivo all'atto dell'autorizzazione, il tenore di sostanze attive e la data limite di garanzia del tenore o la durata di conservazione a decorrere dalla data di fabbricazione; [3]
6) oligoelementi: il nome specifico dell'additivo all'atto dell'autorizzazione e il tenore dei rispettivi elementi, purché all'atto dell'autorizzazione dell'additivo sia fissato un tenore massimo per i mangimi completi; [4]
7) conservanti: il nome specifico dell'additivo all'atto dell'autorizzazione e il tenore di sostanze attive, purché all'atto dell'autorizzazione dell'additivo sia fissato un tenore massimo per i mangimi completi; [5]
8) enzimi: il nome specifico del componente o dei componenti attivi secondo la sua o le loro attività enzimatiche in base all'autorizzazione concessa; il numero di identificazione secondo l'International Union of Biochemisty; le unità di attività (unità di attività per g o per ml); il numero di registrazione CE dell'additivo; il nome o la ragione sociale e l'indirizzo o la sede sociale del fabbricante, se quest'ultimo non è responsabile dell'indicazione di etichettatura; la data limite di garanzia o la durata di conservazione a decorrere dalla data di fabbricazione; il numero di riferimento e la data di fabbricazione della partita; le istruzioni per l'uso che precisano segnatamente la dose raccomandata se necessario con l'indicazione di un minimo e di un massimo in funzione delle percentuali in peso delle materie prime bersaglio per chilogrammo di mangime completo secondo le prescrizioni previste caso per caso nelle autorizzazioni dell'additivo; eventualmente, l'indicazione delle particolari caratteristiche significative dovute al processo di fabbricazione, in base alle disposizioni previste in materia di etichettatura nell'autorizzazione dell'additivo; [6]
9) microrganismi: l'identificazione del ceppo in base all'autorizzazione concessa, il numero di deposito del ceppo, il numero di unità che formano colonie (CFU/g), il numero di registrazione CE dell'additivo, il nome o la ragione sociale e l'indirizzo o la sede sociale del fabbricante, se quest'ultimo non è responsabile delle indicazioni di etichettatura, la data limite di garanzia o la durata di conservazione a decorrere dalla data di fabbricazione, l'eventuale indicazione delle particolari caratteristiche significative derivanti dal processo di fabbricazione, in base alle disposizioni previste in materia di etichettatura nell'autorizzazione dell'additivo; [7]
10) altri additivi appartenenti ai gruppi di cui ai punti da 2) a 9), per i quali non è previsto alcun tenore massimo, e additivi appartenenti ad altri gruppi autorizzati: il nome specifico attribuito all'additivo all'atto dell'autorizzazione dell'additivo e il tenore di sostanze attive, purché tali additivi abbiano una funzione a livello dei mangimi e siano dosabili con metodi di analisi ufficiali o, in loro mancanza, con metodi scientificamente validi. [8]
2. Il nome specifico degli additivi può essere accompagnato dalla denominazione commerciale.
3. Il nome del produttore di additivi di cui al comma 1, lettera b), numero 1), deve essere indicato sull'etichetta delle premiscele.
4. Il nome specifico degli additivi autorizzati può essere accompagnato dal numero di registrazione CE dell'additivo.
5. Qualora, conformemente al comma 1, debba essere dichiarata la data limite di garanzia o la durata di conservazione a decorrere dalla data di fabbricazione di vari additivi appartenenti ad uno stesso gruppo o a gruppi diversi, può essere indicata per l'insieme degli additivi una sola data di garanzia o una sola durata di conservazione, purché sia quella che scade per prima.
6. Gli imballaggi, i recipienti e le etichette possono contenere indicazioni diverse da quelle prescritte o ammesse ai sensi dei commi da 1 a 5, purché esse siano nettamente separate dalle indicazioni di cui sopra.
7. Il produttore, il condizionatore, l'importatore, il venditore o il distributore stabiliti nel territorio comunitario sono responsabili dell'applicazione delle disposizioni del presente articolo.
8. Per la commercializzazione fra gli Stati membri le indicazioni di cui al presente articolo saranno redatte almeno in una delle lingue ufficiali del Paese destinatario; per la commercializzazione in Italia, tali indicazioni devono essere redatte in lingua italiana.
Art. 18. Etichettatura dei mangimi
1. I mangimi nei quali sono stati incorporati gli additivi appartenenti ai gruppi sotto elencati possono essere immessi in circolazione soltanto se figurano, sull'imballaggio, sul recipiente o sull'etichetta ivi fissata in modo ben visibile, chiaramente leggibile ed indelebile, le seguenti indicazioni:
a) per gli antibiotici, i coccidiostatici e le altre sostanze medicamentose, nonché per i fattori di crescita: nome specifico attribuito all'additivo all'atto dell'autorizzazione, tenore di sostanze attive e data limite di garanzia del tenore o durata di conservazione a decorrere dalla data di fabbricazione, numero di riconoscimento attribuito all'impresa ai sensi dell'articolo 4 del citato
b) per le sostanze che hanno effetti antiossidanti:
1) mangimi per animali familiari: l'indicazione "con antiossidante" seguita dal nome specifico attribuito all'additivo all'atto dell'autorizzazione;
2) mangimi composti diversi da quelli destinati agli animali familiari: nome specifico attribuito all'additivo all'atto dell'autorizzazione;
c) per le sostanze coloranti, compresi i pigmenti, purché siano utilizzati per la colorazione del mangime o dei prodotti animali:
1) mangimi per animali familiari: l'indicazione "coloranti" o "colorato con" seguita dal nome specifico attribuito all'additivo all'atto dell'autorizzazione;
2) mangimi composti diversi da quelli destinati agli animali familiari: nome specifico attribuito all'additivo all'atto dell'autorizzazione;
d) per la vitamina E: nome specifico attribuito all'additivo all'atto dell'autorizzazione, tenore di alfa-tocoferoli e data limite di garanzia del tenore o durata di conservazione a decorrere dalla data di fabbricazione;
e) per le vitamine A e D: nome specifico attribuito all'additivo all'atto dell'autorizzazione, tenore di sostanze attive e data limite di garanzia del tenore o durata di conservazione a decorrere dalla data di fabbricazione;
f) per il rame: nome specifico attribuito all'additivo all'atto dell'autorizzazione e tenore espresso in rame (Cu);
g) per i conservanti:
1) mangimi per animali familiari: l'indicazione "conservante" o "conservato con" seguita dal nome specifico attribuito all'additivo all'atto dell'autorizzazione;
2) mangimi composti diversi da quelli destinati agli animali familiari: nome specifico attribuito all'additivo all'atto dell'autorizzazione;
h) per gli enzimi: il nome specifico del o dei composti attivi secondo la sua o le loro attività enzimatiche, in base all'autorizzazione concessa, il numero di identificazione secondo l'International Union of Biochemistry, le unità di attività espresse per chilogrammo o per litro, il numero di registrazione CE dell'additivo, la data limite di garanzia o la durata di conservazione a decorrere dalla data di fabbricazione, l'eventuale indicazione delle caratteristiche particolari significative derivanti dal processo di fabbricazione, in base alle disposizioni previste in materia di etichettatura nell'autorizzazione dell'additivo;
i) per i microrganismi: l'identificazione del ceppo in base all'autorizzazione concessa, il numero di deposito del ceppo, il numero di unità che formano colonie (CFU per chilogrammo), il numero di registrazione CE dell'additivo, la data limite di garanzia o la durata di conservazione a decorrere dalla data di fabbricazione, eventualmente l'indicazione delle caratteristiche particolari significative dovute al processo di fabbricazione, in base alle disposizioni previste in materia di etichettatura nell'autorizzazione dell'additivo.
2. Oltre a quelle di cui al comma 1, eventuali altre indicazioni, in particolare quelle relative all'utilizzazione appropriata del mangime, prescritte dall'autorizzazione comunitaria dell'additivo, debbono figurare sull'imballaggio, sul recipiente o su un'etichetta ad essi fissata.
3. La presenza di oligoelementi diversi dal rame nonché la presenza di vitamine, diverse dalle vitamine A, D ed E, di provitamine e di sostanze aventi un effetto analogo può essere indicata soltanto quando tali additivi sono dosabili secondo metodi ufficiali di analisi o, in mancanza, secondo metodi scientificamente riconosciuti. In tal caso devono essere fornite le seguenti indicazioni:
a) per gli oligoelementi diversi dal rame: nome specifico dell'additivo risultante dall'autorizzazione e tenore dei rispettivi elementi;
b) per le vitamine diverse dalle vitamine A, D ed E, le provitamine e le sostanze aventi un effetto analogo: nome specifico dell'additivo risultante dall'autorizzazione, tenore di sostanze attive e data limite di garanzia del tenore o durata di conservazione a decorrere dalla data di fabbricazione.
4. Le indicazioni di cui ai commi 1, 2 e 3, devono figurare accanto a quelle riportate sull'imballaggio, sul recipiente o su un'etichetta ivi fissata a norma delle vigenti disposizioni in materia di mangimi.
5. Qualora, conformemente ai commi 1, 2 e 3, si dichiari un tenore o una quantità, tale dichiarazione deve riferirsi alla parte di additivo incorporata nel mangime.
6. La indicazione degli additivi di cui ai commi 1, 2 e 3, deve essere accompagnata dal numero di registrazione CE dell'additivo o dalla denominazione commerciale, nel caso in cui tali indicazioni non siano richieste ai sensi del comma 1.
7. Qualora, ai sensi del comma 1, debba essere dichiarata la data limite di garanzia o la durata di conservazione a decorrere dalla data di fabbricazione di vari additivi appartenenti ad uno stesso gruppo o a gruppi diversi, può essere indicata per gli insiemi degli additivi una sola data di garanzia o una sola durata di conservazione a decorrere dalla data di fabbricazione, e, precisamente, quella che scade per prima.
8. Nel caso di mangimi commercializzati in autocisterne, veicoli analoghi o alla rinfusa, le indicazioni di cui ai commi 1, 2 e 3, figurano su un documento di accompagnamento. Qualora si tratti di piccole quantità di mangimi destinati all'utilizzazione finale, è sufficiente che le indicazioni siano portate a conoscenza dell'acquirente, con le modalità di cui all'articolo 18, comma 10, della
9. Nel caso di mangimi per animali familiari contenenti coloranti, conservanti o sostanze aventi effetti antiossidanti, condizionati in imballaggi con un contenuto netto di peso pari o inferiore a 10 Kg, è sufficiente che l'imballaggio rechi rispettivamente l'indicazione "colorante", "colorato con", "conservato con", "con antiossidante" seguita dalle parole "additivi CE", a condizione che sull'imballaggio, sul recipiente o sull'etichetta sia indicato un numero di riferimento che consenta l'identificazione del mangime e che, su richiesta, il fabbricante comunichi il nome specifico dell'additivo o degli additivi utilizzati.
10. E' vietata qualsiasi indicazione relativa agli additivi diversa da quelle prevista nel presente decreto.
11. Per la commercializzazione fra gli Stati membri le indicazioni di cui al presente articolo saranno redatte almeno in una delle lingue ufficiali del Paese destinatario; per la commercializzazione in Italia, tali indicazioni devono essere redatte in lingua italiana.
12. Il produttore, il condizionatore, l'importatore, il venditore o il distributore stabiliti nel territorio comunitario sono responsabili dell'applicazione delle disposizioni del presente articolo.
Art. 19. Ulteriori norme sui mangimi complementari
1. Fatte salve le disposizioni di cui alla
2. La disposizione di cui al comma 1 non si applica ai prodotti consegnati ai fabbricanti di mangimi composti o ai loro fornitori.
3. La dichiarazione di cui al comma 1 è redatta in modo che, nel caso di utilizzazione conforme, la proporzione degli additivi non superi il tenore massimo fissato per i mangimi completi.
Art. 20. Vigilanza e controlli
1. Le autorità competenti, effettuano, nel corso della commercializzazione, almeno a campione, ai sensi del
2. In mancanza di disposizioni comunitarie che fissano le tolleranze in caso di divergenza tra il risultato del controllo ufficiale ed il tenore dichiarato dell'additivo nel mangime composto, le tolleranze stesse sono stabilite secondo la procedura di cui all'articolo 24, comma 3, della
3. In caso di interazione indesiderabile e imprevista tra gli additivi di cui all'articolo 2, comma 1, lettera g), ed altri additivi o prodotti medicinali veterinari, il responsabile dell'immissione in circolazione dell'additivo, o il suo rappresentante stabilito nel territorio comunitario, in caso di additivi originari da Paesi terzi, raccoglie tutte le informazioni inerenti e le trasmette al Ministero della salute.
Art. 21. Norme transitorie
1. Dalla data di entrata in vigore del presente regolamento è consentito un periodo di sei mesi per lo smaltimento delle giacenze degli additivi delle premiscele e dei mangimi che li contengono e delle etichette, imballaggi e confezioni conformi alla normativa previgente.
Art. 22. Disposizioni finanziarie
1. Le spese relative alle attività previste dall'articolo 4, comma 1, dall'articolo 6, commi 2 e 5, dall'articolo 7, comma 5, e dall'articolo 8, comma 3, sono a carico del richiedente.
2. Con decreto del Ministro della salute, di concerto con il Ministro dell'economia e delle finanze, da emanarsi entro novanta giorni dalla data di entrata in vigore del presente regolamento, sono determinate ed aggiornate ogni due anni, sulla base dei criteri stabiliti dall'allegato B della decisione 98/728/CE, le tariffe derivanti dall'applicazione del presente articolo nonché le relative modalità di versamento.
Art. 23. Abrogazioni
1. Il decreto del Presidente della Repubblica 1° marzo 1992, n. 228, e successive modificazioni, è abrogato.Il presente decreto, munito del sigillo dello Stato, sarà inserito nella Raccolta ufficiale degli atti normativi della Repubblica italiana. E' fatto obbligo a chiunque spetti di osservarlo e di farlo osservare.
Allegato A
Parte prima:
antibiotici: tutti gli additivi del gruppo;
coccidiostatici e altre sostanze medicamentose: tutti gli additivi del gruppo;
fattori di crescita: tutti gli additivi del gruppo.
Parte seconda:
oligoelementi: rame e selenio;
vitamine, provitamine e sostanze con effetto analogo chimicamente ben definite: vitamine A e D.
Allegato B
Elenco degli additivi associati al responsabile dell'immissione in circolazione iscritti nell'allegato I anteriormente al 1° gennaio 1988
|
A. ANTIBIOTICI |
||||
|
Numero di |
Nome del |
Additivo |
|
|
|
registrazione |
responsabile |
|
Designazione chimica |
Specie animale o categoria di |
|
|
dell'immissione in |
|
descrizione |
animali |
|
|
circolazione (*) |
|
|
|
|
E 712 |
|
Flavofosfolipol |
C70H124O40N6P |
Galline ovaiole |
|
|
|
|
|
Tacchini |
|
|
|
|
|
Altro pollame ad eccezione delle |
|
|
|
|
|
anatre, oche e piccioni |
|
|
|
|
|
Suinetti |
|
|
|
|
|
Suini |
|
|
|
|
|
Animali da pelliccia ad eccezione |
|
|
|
|
|
dei conigli |
|
|
|
|
|
Vitelli |
|
|
|
|
|
|
|
|
|
|
|
Bovini da ingrasso |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
E 714 |
|
Monensin sodico |
C36H61O11Na (Sale |
Bovini da ingrasso |
|
|
|
|
sodico del polietere dell'acido |
|
|
|
|
|
monocarbossilico prodotto da |
|
|
|
|
|
Streptomyces cinnamonensis) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(*) Autorizzazione da associare al responsabile a decorrere al 1° ottobre 1999. |
||||
|
|
||||
|
Numero di |
|
Tenore |
Tenore |
|
|
registrazione |
Età massima |
minimo |
massimo |
Altre disposizioni |
|
|
|
|
|
|
|
|
|
mg/Kg di mangime completo |
|
|
|
E 712 |
- |
2 |
5 |
- |
|
|
26 settimane |
1 |
20 |
- |
|
|
16 settimane |
1 |
20 |
- |
|
|
|
|
|
|
|
|
3 mesi |
10 |
25 |
Solo negli alimenti per allevamento |
|
|
6 mesi |
1 |
20 |
- |
|
|
- |
2 |
4 |
- |
|
|
|
|
|
|
|
|
6 mesi |
6 |
16 |
- |
|
|
6 mesi |
8 |
16 |
Solo negli alimenti per allattamento |
|
|
- |
2 |
10 |
Indicazioni che devono figurare nel |
|
|
|
|
|
modo di impiego: "per gli alimenti |
|
|
|
|
|
complementari la dose massima |
|
|
|
|
|
nella razione giornaliera non deve |
|
|
|
|
|
superare: per 100 kg di peso animale: |
|
|
|
|
|
40 mg; oltre i 100 kg aggiungere 1,5 |
|
|
|
|
|
mg per ogni 10 ulteriori chili di peso |
|
|
|
|
|
animale" |
|
E 714 |
- |
10 |
40 |
Indicazioni che devono figurare nel |
|
|
|
|
|
modo di impiego: |
|
|
|
|
|
"per gli alimenti complementari la |
|
|
|
|
|
dose massima nella razione |
|
|
|
|
|
giornaliera non deve superare: per |
|
|
|
|
|
100 kg di peso animale: 140 mg; oltre |
|
|
|
|
|
i 100 kg: aggiungere 6 mg per ogni 10 |
|
|
|
|
|
ulteriori kg di peso animale". |
|
|
|
|
|
"Pericoloso per gli equidi" |
|
|
|
|
|
"Questo alimento contiene un additivo |
|
|
|
|
|
del gruppo degli ionofori: la sua |
|
|
|
|
|
somministrazione contemporanea a |
|
|
|
|
|
taluni medicinali (ad esempio la |
|
|
|
|
|
tiamulina) può essere controindicata". |
|
(*) Autorizzazione da associare al responsabile a decorrere dal 1° ottobre 1999. |
||||
|
B. COCCIDIOSTATICI E ALTRE SOSTANZA MEDICAMENTOSE |
|||||
|
Numero di |
Nome del |
|
|
|
|
|
registrazione |
responsabile |
Additivo |
Designazione chimica |
Specie animale o categoria di |
|
|
|
dell'immissione in |
|
descrizione |
animali |
|
|
|
circolazione (*) |
|
|
|
|
|
E 750 |
|
Amprolium |
Cloridrato del cloruro di 1- |
Pollame |
|
|
|
|
|
[(4-amino-2propil5-piriminidil) |
|
|
|
|
|
|
metil]-picolinio |
|
|
|
|
|
|
|
|
|
|
E 751 |
|
Amprolium-etopabato |
(a) Cloridrato del cloruro di 1 |
Polli, tacchini e faraone |
|
|
|
|
(miscela: 25 parti di a) |
[(4-animo-2-propil-5-piriminidil) |
|
|
|
|
|
amprolium e 1,6 parti |
metil]-picolinio |
|
|
|
|
|
di b) etopabato |
|
|
|
|
|
|
(b) |
|
|
|
|
|
|
Metil-4-acetamide-2- e- |
|
|
|
|
|
|
tossibenzoato |
|
|
|
|
E 754 |
|
Dimetridazolo |
1,2-dimetil-5-nitroimidazolo |
Tacchini |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Faraone |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
E 755 |
|
Meticlorpindolo |
3,5-dicloro-2,6-dimetil-4-piridin |
Polli da ingrasso, faraone |
|
|
|
|
|
lo |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Conigli |
|
|
|
|
|
|
|
|
|
E 756 |
|
Decochinato |
3-Etossicarbonil-4-idrossi-6- |
Polli da ingrasso |
|
|
|
|
|
decillosi-7-etossi-chinoleina |
|
|
|
E 757 |
|
Monensin sodico |
C36H61O11Na (Sale |
Polli da ingrasso |
|
|
|
|
|
sodico del polietere dell'acido |
|
|
|
|
|
|
monocarbossilico prodotto da |
|
|
|
|
|
|
Streptomyces cinnamonensis) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Pollastre destinate alla produzione |
|
|
|
|
|
|
di uova |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Tacchini |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
E 758 |
|
Robenidina |
Cloridrato di 1,3-bis |
Polli da ingrasso, tacchini |
|
|
|
|
|
[(4-clorobenzilidene)amino]- |
|
|
|
|
|
|
guanidina |
|
|
|
|
|
|
|
Conigli da ingrasso |
|
|
|
|
|
|
|
|
|
E 761 |
|
Meticlorpindolo/Metil- |
(a) 3,5.dicloro-2,6-dimetil-4- |
Polli da ingrasso |
|
|
|
|
benzoquato parti di (a) |
piridinolo |
|
|
|
|
|
(Miscela 100 |
(b) |
Pollastre destinate alla produzione |
|
|
|
|
meticlorpindolo e 8,35 |
7-Benzilossi-6-butil-3-metossi |
di uova |
|
|
|
|
parti di (b) |
carbonil-4-chinolone |
Tacchini |
|
|
|
|
metilbenzoquato |
|
|
|
|
E 763 |
|
Lasalocid sodico |
C34H53O8Na (sale |
Polli da ingrasso |
|
|
|
|
|
iodico del polietere dell'acido |
|
|
|
|
|
|
monocarbamilico prodotto da |
|
|
|
|
|
|
Streptomyces lasaliensis) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Pollastre destinate alla produzione |
|
|
|
|
|
|
di uova |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
E 764 |
|
Alofuginone |
4(311)-Chinazolinone-7-bromo- |
Polli da ingrasso |
|
|
|
|
|
6-cloro-[3-(3-idrossi-2-piperidil) |
|
|
|
|
|
|
acetonil]-dl-transbromidrato |
|
|
|
|
|
|
|
Tacchini |
|
|
|
|
|
|
|
|
|
E 765 |
|
Narasin |
C43H72O11(Polietere |
Polli da ingrasso |
|
|
|
|
|
dell'acido monocarbossilico |
|
|
|
|
|
|
prodotto da Streptomyces |
|
|
|
|
|
|
aureofaciens) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
E 766 |
|
Salinomicina sodica |
C42H69O11Na (Sale |
Polli da ingrasso |
|
|
|
|
|
sodico del polietere dell'acido |
|
|
|
|
|
|
monocarbossilico prodotto da |
|
|
|
|
|
|
Streptomyces albus) |
|
|
|
|
|
|
Tenore in Elaiofilina: meno di |
|
|
|
|
|
|
42 mg per Kg di |
|
|
|
|
|
|
Salinomicina-sodica |
|
|
|
|
|
|
Tenore in |
|
|
|
|
|
|
17-epi-20-desossisalinomicina: |
|
|
|
|
|
|
meno di 40 g per Kg |
|
|
|
|
|
|
di Salinomicina-sodica |
|
|
|
E 768 |
|
Nicarbazina |
Complessoequimolecolare di |
Polli da ingrasso |
|
|
|
|
|
1,3-bis(4-nitrofenil)urea e di |
|
|
|
|
|
|
4,6-dimetil-2-pirimidinolo |
|
|
|
(*) Autorizzazione da associare al responsabile a decorrere dal 1° ottobre 1999. |
|||||
|
|
|||||
|
Numero di |
|
Tenore |
Tenore |
|
|
|
registrazione |
Età massima |
minimo |
massimo |
Altre disposizioni |
|
|
|
|
|
|
|
|
|
|
|
mg/Kg di mangime completo |
|
||
|
E 750 |
- |
62,5 |
125 |
Somministrazione vietata deposizione |
|
|
|
|
|
|
rispettivamente a partire dall'età di |
|
|
|
|
|
|
e almeno tre giorni prima della |
|
|
|
|
|
|
macellazione |
|
|
E 751 |
- |
66,5 |
133 |
Somministrazione vietata deposizione |
|
|
|
|
|
|
rispettivamente a partire dall'età di |
|
|
|
|
|
|
e almeno tre giorni prima della |
|
|
|
|
|
|
macellazione |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
E 754 |
- |
100 |
200 |
Somministrazione vietata deposizione |
|
|
|
|
|
|
rispettivamente a partire dall'età di |
|
|
|
|
|
|
e almeno sei giorni prima della |
|
|
|
|
|
|
macellazione |
|
|
|
- |
125 |
150 |
Somministrazione vietata deposizione |
|
|
|
|
|
|
rispettivamente a partire dall'età di |
|
|
|
|
|
|
e almeno sei giorni prima della |
|
|
|
|
|
|
macellazione |
|
|
E 755 |
- |
125 |
125 |
Somministrazione vietata a partire |
|
|
|
|
|
|
dall'età di deposizione e almeno |
|
|
|
|
|
|
cinque giorni prima della macellazione |
|
|
|
- |
125 |
200 |
Somministrazione vietata almeno |
|
|
|
|
|
|
cinque giorni prima della macellazione |
|
|
E 756 |
- |
20 |
40 |
Somministrazione vietata almeno tre |
|
|
|
|
|
|
giorni prima della macellazione |
|
|
E 757 |
- |
100 |
125 |
Somministrazione vietata almeno tre |
|
|
|
|
|
|
giorni prima della macellazione. |
|
|
|
|
|
|
Indicare nel modo di impiego |
|
|
|
|
|
|
"pericoloso per gli equidi". "Questo |
|
|
|
|
|
|
alimento contiene un additivo del |
|
|
|
|
|
|
gruppo degli ionofori: la sua |
|
|
|
|
|
|
somministrazione contemporanea a |
|
|
|
|
|
|
taluni medicinali (ad esempio la |
|
|
|
|
|
|
tiamulina) può essere controindicata" |
|
|
|
16 settimane |
100 |
120 |
Indicare nel modo di impiego alimento |
|
|
|
|
|
|
"pericoloso per gli equidi". "Questo |
|
|
|
|
|
|
contiene un additivo del gruppo degli |
|
|
|
|
|
|
ionofori: la sua somministrazione |
|
|
|
|
|
|
contemporanea a taluni medicinali (ad |
|
|
|
|
|
|
esempio la tiamulina) può essere |
|
|
|
|
|
|
controindicata" |
|
|
|
16 settimane |
90 |
100 |
Somministrazione vietata almeno tre |
|
|
|
|
|
|
giorni prima della macellazione. |
|
|
|
|
|
|
Indicare nel modo di impiego: |
|
|
|
|
|
|
"pericoloso per gli equidi". "Questo |
|
|
|
|
|
|
alimento contiene un additivo del |
|
|
|
|
|
|
gruppo degli ionofori: la sua |
|
|
|
|
|
|
somministrazione contemporanea a |
|
|
|
|
|
|
taluni medicinali (ad esempio la |
|
|
|
|
|
|
tiamulina) può essere controindicata". |
|
|
E 758 |
- |
30 |
36 |
Somministrazione vietata almeno |
|
|
|
|
|
|
cinque giorni prima della macellazione |
|
|
|
|
|
|
|
|
|
|
- |
50 |
66 |
Somministrazione vietata almeno tre |
|
|
|
|
|
|
giorni prima della macellazione. |
|
|
E 761 |
- |
110 |
110 |
Somministrazione vietata almeno |
|
|
|
|
|
|
cinque giorni prima della macellazione |
|
|
|
16 settimane |
110 |
110 |
- |
|
|
|
|
|
|
|
|
|
|
12 settimane |
110 |
110 |
Somministrazione vietata almeno |
|
|
|
|
|
|
cinque giorni prima della macellazione |
|
|
E 763 |
- |
75 |
125 |
Somministrazione vietata almeno |
|
|
|
|
|
|
cinque giorni prima della |
|
|
|
|
|
|
macellazione. |
|
|
|
|
|
|
Indicare nel modo di impiego: "Questo |
|
|
|
|
|
|
alimento contiene un additivo del |
|
|
|
|
|
|
gruppo degli ionofori: la sua |
|
|
|
|
|
|
somministrazione contemporanea a |
|
|
|
|
|
|
taluni medicinali può essere |
|
|
|
|
|
|
controindicata" |
|
|
|
16 settimane |
75 |
125 |
Indicare nel modo di impiego: "Questo |
|
|
|
|
|
|
alimento contiene un additivo del |
|
|
|
|
|
|
gruppo degli ionofori: la sua |
|
|
|
|
|
|
somministrazione contemporanea a |
|
|
|
|
|
|
taluni medicinali può essere |
|
|
|
|
|
|
controindicata“ |
|
|
E 764 |
- |
2 |
3 |
Somministrazione vietata almeno |
|
|
|
|
|
|
cinque giorni prima della macellazione |
|
|
|
|
|
|
|
|
|
|
12 settimane |
2 |
3 |
Somministrazione vietata almeno |
|
|
|
|
|
|
cinque giorni prima della macellazione |
|
|
E 765 |
- |
60 |
70 |
Somministrazione vietata almeno |
|
|
|
|
|
|
cinque giorni prima della |
|
|
|
|
|
|
macellazione. |
|
|
|
|
|
|
Indicare nel modo di impiego : |
|
|
|
|
|
|
"Pericoloso per gli equidi“. |
|
|
|
|
|
|
"Questo alimento contiene un additivo |
|
|
|
|
|
|
del gruppo degli ionofori: la sua |
|
|
|
|
|
|
somministrazione contemporanea a |
|
|
|
|
|
|
taluni medicinali può essere |
|
|
|
|
|
|
controindicata" |
|
|
E 766 |
- |
50 |
70 |
Somministrazione vietata almeno |
|
|
|
|
|
|
cinque giorni prima della macellazione |
|
|
|
|
|
|
Indicare nel modo di impiego: |
|
|
|
|
|
|
"Pericoloso per gli equidi" |
|
|
|
|
|
|
"Questo alimento contiene un additivo |
|
|
|
|
|
|
del gruppo degli ionofori: la sua |
|
|
|
|
|
|
somministrazione contemporanea a |
|
|
|
|
|
|
taluni medicinali può essere |
|
|
|
|
|
|
controindicata" |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
E 768 |
4 settimane |
100 |
125 |
Somministrazione vietata almeno nove |
|
|
|
|
|
|
giorni prima della macellazione |
|
|
|
|
|
|
|
|
|
(*) Autorizzazione da associare al responsabile a decorrere dal 1° ottobre 1999. |
|||||
Allegato C
PARTE I
Additivi che sono oggetto di un'autorizzazione associata al responsabile della loro immissione in circolazione previsti all'art. 2, comma 1, lettera h):
antibiotici: tutti gli additivi del gruppo;
coccidiostatici e altre sostanze medicamentose: tutti gli additivi del gruppo;
fattori di crescita: tutti gli additivi del gruppo.
PARTE II
Altri additivi di cui all'art. 2, comma 1, lettera g):
sostanze con effetti antiossidanti: tutti gli additivi del gruppo;
sostanze aromatizzanti e aperitive;
emulsionanti, stabilizzanti, addensanti e gelificanti: tutti gli additivi del gruppo;
coloranti compresi i pigmenti: tutti gli additivi del gruppo;
conservanti;
vitamine, provitamine e sostanze con effetto analogo chimicamente ben definite: tutti gli additivi del gruppo;
oligoelementi: tutti gli additivi del gruppo;
agenti leganti, antiagglomeranti e coagulanti: tutti gli additivi del gruppo;
regolatori dell'acidità: tutti gli additivi del gruppo;
enzimi: tutti gli additivi del gruppo;
microrganismi: tutti gli additivi del gruppo.
Allegato D [9]
Parte I
Additivi diversi dai microrganismi e dagli enzimi considerazioni generali
Il presente documento è destinato a costituire una guida per l'istruzione dei fascicoli relativi alle sostanze ed ai preparati per i quali venga richiesta l'autorizzazione quali additivi negli alimenti per animali oppure per quelli relativi ad un nuovo utilizzo di un additivo autorizzato. In queste linee direttrici, il termine "additivo" si riferisce alle sostanze attive chimicamente specificate o ai preparati contenenti principi attivi nello stato in cui essi saranno incorporati nelle premiscele e negli alimenti per animali. I fascicoli devono consentire una valutazione degli additivi sulla base delle conoscenze attuali e garantire che essi rispettino i principi fondamentali fissati per la loro autorizzazione, che costituiscono l'oggetto delle disposizioni di cui all'art. 3 di questo regolamento. Quando un fascicolo riguarda un additivo costituito da o contenente organismi geneticamente modificati di cui al
I fascicoli devono contenere relazioni dettagliate su tutti gli studi condotti, da presentare nell'ordine e con la numerazione proposti nelle presenti linee direttrici. Essi devono comprendere i riferimenti e le copie di tutti i dati scientifici pubblicati che risultino pertinenti alla valutazione dell'additivo. Deve, preferibilmente, essere fornita anche una versione elettronica del fascicolo.
Gli studi sono destinati a dimostrare la sicurezza dell'utilizzo dell'additivo:
a) per la specie bersaglio ai livelli ai quali si propone di incorporare l'additivo nell'alimento per animali;
b) per le persone che potrebbero essere esposte all'additivo per inalazione o altro contatto con mucose, occhi o pelle nel corso della manipolazione dell'additivo in quanto tale o incorporato nelle premiscele o negli alimenti per animali;
c) per i consumatori che ingeriscono prodotti alimentari ottenuti da animali cui sia stato somministrato l'additivo, che potrebbero contenere residui dell'additivo o suoi metaboliti; ciò sarà garantito dalla fissazione dei limiti massimi di residui (MRL) e dei periodi di sospensione;
d) per gli animali e gli esseri umani attraverso la selezione e la diffusione di geni di resistenza antimicrobica;
e) per l'ambiente in relazione all'additivo stesso od ai prodotti da esso derivati direttamente e/o per escrezione da parte degli animali.
In generale devono essere forniti studi per stabilire l'identità, le condizioni di impiego, le proprietà fisicochimiche, i metodi di determinazione e l'efficacia dell'additivo, come pure il suo destino metabolico, i suoi residui e i residui dei prodotti del suo metabolismo, nonché gli effetti biologici e tossicologici sulle specie bersaglio. Quando l'additivo è destinato ad una categoria di animali che appartiene ad una specie determinata, gli studi sull'efficacia e sui residui devono essere condotti su quella categoria bersaglio. Gli studi necessari alla valutazione dei rischi per la salute umana o per l'ambiente dipenderanno essenzialmente dalla natura dell'additivo e dalle circostanze del suo utilizzo. A questo proposito non è applicabile alcuna norma rigida. Se necessario verranno richieste informazioni aggiuntive. L'omissione dal fascicolo di qualsiasi dato richiesto in base alle presenti linee direttrici deve essere motivata. In particolare, gli studi di mutagenicità, di cancerogenicità, di tossicità riproduttiva possono essere evitati solo se la composizione chimica, l'esperienza pratica, le conoscenze scientifiche o altre considerazioni possano far ragionevolmente escludere tali effetti. Gli studi devono essere condotti e le relative relazioni redatte in base ad idonee norme di qualità, ad esempio seguendo le norme di buone prassi di laboratorio (BPL) a norma del decreto ministeriale 26 giugno 1986 e del
Per quanto concerne le relazioni sulla qualità, sull'efficacia e sulla sicurezza devono essere fornite relazioni di esperti, che devono possedere qualifiche pertinenti e documentare una chiara esperienza nel settore specifico. Essi, inoltre non devono aver preso personalmente parte allo svolgimento dei test compresi nel fascicolo. Le relazioni devono contenere una dettagliata valutazione critica della documentazione fornita dal richiedente.
La determinazione delle proprietà fisicochimiche, tossicologiche ed ecotossicologiche deve essere effettuata conformemente ai metodi stabiliti dal
Ciascun fascicolo deve contenere un riepilogo adeguato, una proposta in allegato e può contenere una monografia. I fascicoli relativi agli antibiotici, ai coccidiostatici e ad altre sostanze medicamentose, nonché ai fattori di crescita devono essere accompagnati da una monografia conforme al modello di cui al Capitolo V, che consenta l'identificazione e caratterizzazione dell'additivo a norma degli articoli 3 e 4 del presente regolamento (
Per quanto concerne gli additivi destinati esclusivamente agli alimenti per animali da compagnia può non essere sempre necessario sottoporli ad un programma di test sulla tossicità cronica, sulla mutagenicità, sulla tossicità riproduttiva completo come quello previsto per gli additivi destinati all'alimentazione di animali da reddito dai quali vengono ottenuti prodotti per il consumo umano. Non sono richiesti studi sui residui negli animali da compagnia.
Per le finalità di seguito indicate è richiesto lo studio del metabolismo dell'additivo negli animali bersaglio da cui vengono ricavate derrate alimentari umane e nelle specie di laboratorio utilizzate per le prove di tossicità:
a) garantire la disponibilità di dati sufficienti sulla tossicità dell'additivo progenitore e degli eventuali metaboliti prodotti nella specie bersaglio cui il consumatore potrebbe essere esposto. A tal fine è importante un confronto tra il destino metabolico dell'additivo nelle specie animali bersaglio e in quelle di laboratorio utilizzate per le prove di tossicità.
b) individuare e quantificare gli opportuni residui marcatori da utilizzare per fissare l'MRL del residuo marcatore ed i periodi di sospensione per il prodotto finale.
CAPITOLO I: Riepilogo dei dati nel fascicolo.
Il riepilogo deve seguire l'ordine delle linee direttrici e trattare tutte le varie parti con indicazione dei riferimenti alle pagine corrispondenti del fascicolo. Deve, inoltre, contenere una proposta che rispetti tutte le condizioni previste per l'autorizzazione richiesta.
Capitolo II: Identità, caratteristiche e condizioni d'impiego dell'additivo; metodi di controllo.
II.1. Identità dell'additivo.
1.1. Denominazioni commerciali previste per la commercializzazione
1.2. Tipo di additivo in base all'azione principale. Laddove possibile, devono essere allegate prove relative ai suoi meccanismi di azione. Eventuali altri impieghi del principio attivo devono essere precisati.
1.3. Composizione qualitativa e quantitativa (principio attivo, altri componenti, impurità, variazione tra le partite). Se il principio attivo è costituito da una miscela di vari componenti attivi ciascuno dei quali chiaramente definibile, occorre descrivere separatamente i principali componenti e indicare le loro proporzioni nella miscela.
1.4. Stato fisico, distribuzione delle particelle in base alla loro dimensione, forma delle particelle, densità, peso specifico apparente; per i liquidi: viscosità, tensione superficiale.
1.5. Procedimento di fabbricazione, compresi eventuali trattamenti specifici.
II.2. Caratterizzazione delle sostanze attive.
2.1. Denominazione generica, denominazione chimica secondo la nomenclatura UICPA (Unione internazionale di chimica pura e applicata), altre denominazioni generiche internazionali e abbreviazioni. Numero CAS (Chemical Abstracts Service Number).
2.2. Formula di struttura, formula molecolare e peso molecolare.
Se i principi attivi sono prodotti di fermentazione, indicare origine microbica (denominazione e sede della raccolta delle colture riconosciuta quale autorità internazionale di deposito possibilmente nell'Unione europea, presso la quale il ceppo è depositato, numero di deposito del ceppo, nonché tutte le pertinenti caratteristiche morfologiche, fisiologiche, genetiche e molecolari necessarie alla sua identificazione). Per i ceppi geneticamente modificati devono essere fornite informazioni sulla modificazione genetica.
2.3. Purezza
Identificazione e quantificazione delle eventuali impurità chimiche e microbiche nonché delle sostanze tossiche, conferma dell'assenza di organismi produttori.
2.4. Proprietà pertinenti
Proprietà fisiche delle sostanze chimicamente specificate: costante di dissociazione, pKa, proprietà elettrostatiche, punto di fusione, punto di ebollizione, densità, pressione di vapore, solubilità in acqua e in solventi organici, Kow e Koc, spettri di massa e di assorbimento, dati RMN, possibili isomeri ed ogni altra opportuna proprietà fisica.
2.5. Procedimenti di fabbricazione e purificazione, mezzi utilizzati e, nel caso dei prodotti di fermentazione, variazione tra le partite.
II.3. Caratterizzazione dell'additivo: proprietà fisicochimiche e tecnologiche.
3.1. Stabilità di ciascuna formulazione dell'additivo rispetto alle condizioni ambientali, quali luce, temperatura, pH, umidità, ossigeno e materiale di imballaggio. Durata di conservazione dell'additivo nella forma in cui esso viene commercializzato.
3.2. Stabilità di ciascuna formulazione dell'additivo durante la preparazione e la conservazione delle premiscele e degli alimenti per animali, in particolare stabilità rispetto alle previste condizioni di lavorazione/conservazione (calore, umidità, pressione/sollecitazione di taglio, tempo e materiale d'imballaggio). Possibili prodotti di degradazione o decomposizione. Durata di conservazione prevista dell'additivo.
3.3. Altre proprietà fisicochimiche o tecnologiche idonee ad ottenere e conservare miscele omogenee nelle premiscele e negli alimenti per animali, proprietà antipolvere ed elettrostatiche, possibilità di dispersione nei liquidi.
3.4. Incompatibilità o interazioni prevedibili con gli alimenti per animali, gli eccipienti, altri additivi approvati o con medicinali.
II.4. Condizioni di utilizzo dell'additivo.
4.1. Quando un additivo produce notevoli effetti tecnologici e zootecnici deve soddisfare i requisiti previsti per entrambi gli effetti ad esso attribuiti. Devono essere individuati e motivati gli effetti attribuiti a ciascun additivo.
4.2. Utilizzo tecnologico proposto nella produzione di alimenti per animali o, se del caso, nelle materie prime.
4.3. Modalità di utilizzo proposta nell'alimentazione animale (ad esempio specie o categorie di animali e classe di eta/fase di produzione dell'animale, tipo di alimento e controindicazioni).
4.4. Metodo e livello proposti per l'inclusione nelle premiscele e negli alimenti per animali o, se del caso, nelle materie prime, in percentuale ponderale dell'additivo e delle sostanze chimicamente specificate rispetto alle premiscele, agli alimenti per animali o, se del caso, alle materie prime, con proposta della dose nell'alimento finale per animali, proposta della durata di somministrazione e del periodo di sospensione laddove opportuno.
4.5. Devono essere forniti i dati relativi ad altri usi noti del principio attivo (ad esempio in prodotti alimentari, nella medicina umana o veterinaria, in agricoltura e nell'industria).
4.6. Proposta di una scheda di dati di sicurezza dei materiali, come previsto
II.5. Metodi di controllo.
5.1. Descrizione dei metodi utilizzati per la determinazione dei criteri indicati ai punti 1.3, 1.4, 2.3, 2.4, 3.1, 3.2, 3.3 e 3.4 di questo capitolo.
5.2. Descrizione dei metodi analitici qualitativi e quantitativi per il controllo ordinario del principio attivo nelle premiscele e negli alimenti per animali. Tale metodo deve essere convalidato mediante analisi di confronto (ring test) che coinvolgano almeno quattro laboratori oppure deve essere convalidato internamente sulla base di linee direttrici armonizzate a livello internazionale per la convalida interna dei metodi di analisi in relazione ai seguenti parametri: applicabilità, selettività, calibrazione, accuratezza, precisione, campo di variazione, limite di rilevazione, limite di quantificazione, sensibilità, robustezza e fattibilità. Devono essere fornite le prove della valutazione di tali caratteristiche (5.4).
5.3. Descrizione dei metodi analitici qualitativi e quantitativi per la determinazione dei residui marcatori del principio attivo nei tessuti bersaglio e nei prodotti di origine animale.
5.4. I metodi di cui ai punti 5.2 e 5.3 devono essere accompagnati da informazioni concernenti il metodo di campionamento utilizzato, i tassi di recupero, la specificità, l'accuratezza, la precisione, i limiti di rilevazione, i limiti di quantificazione e la procedura di convalida utilizzata. Devono essere forniti i campioni di riferimento del principio attivo e/o dei residui marcatori, nonché informazioni sulle condizioni di conservazione ottimali per questi campioni di riferimento. Nel mettere a punto i metodi, occorre tener conto del fatto che i relativi limiti di quantificazione devono essere inferiori agli MRL. Deve essere inoltre considerato se essi siano idonei per le analisi ordinarie.
Capitolo III: Studi relativi all'efficacia dell'additivo.
III.1. Studi degli effetti sugli alimenti per animali.
Tali studi riguardano additivi tecnologici, quali antiossidanti, conservanti, leganti, emulsionanti, stabilizzanti, gelificanti, modificatori del pH, ecc., che sono destinati a migliorare o stabilizzare le caratteristiche delle premiscele e degli alimenti per animali ma non hanno alcun effetto biologico diretto sulla produzione animale. Tutte le attività o gli effetti attribuiti all'additivo devono essere motivati mediante informazioni scientifiche.
Occorre dimostrare l'efficacia dell'additivo nelle condizioni d'impiego previste mediante il confronto con opportuni mangimi di riferimento, avvalendosi di criteri opportuni rappresentati da metodi accettabili riconosciuti. Le ricerche devono essere concepite e realizzate in modo da consentire una valutazione statistica.
Devono essere fornite informazioni complete sul principi attivi, sui preparati, sulle premiscele e sugli alimenti esaminati, sul numero di riferimento dei lotti, sul trattamento dettagliato e sulle condizioni delle prove. Per ciascun esperimento devono essere descritti gli effetti positivi e negativi d'ordine sia tecnologico sia biologico.
III.2. Studi degli effetti sugli animali.
Gli studi sugli additivi zootecnici devono essere eseguiti nelle specie bersaglio/categorie di animali cui è destinato l'additivo facendo una comparazione con gruppi di controllo negativo (cui non siano somministrati antibiotici, fattori della crescita o altre sostanze medicamentose) ed eventualmente con gruppi di animali alimentati con mangimi contenenti additivi approvati nell'UE e di nota efficacia, somministrati nel dosaggio raccomandato (controllo positivo).
Gli animali utilizzati devono essere sani e preferibilmente appartenere ad un gruppo omogeneo. Gli studi devono consentire la valutazione dell'efficacia dell'additivo in rapporto alle pratiche di allevamento dell'UE. Per tutte le sperimentazioni devono, laddove possibile, essere utilizzati modelli di protocollo simili in modo che in ultima analisi i dati possano essere controllati per verificarne l'omogeneità ed aggregati (qualora tale sia l'indicazione dei test) ai fini della valutazione statistica.
Non viene raccomandato un unico modello; è prevista la flessibilità per lasciare spazio alla libertà scientifica nella progettazione ed esecuzione degli studi. Il modello sperimentale utilizzato deve essere motivato sulla base delle caratteristiche addotte per l'utilizzo dell'additivo e comportare un'adeguata potenza statistica.
2.1. Per i coccidiostatici ed altre sostanze medicamentose
L'importanza deve essere attribuita principalmente alle prove degli effetti specifici (ad esempio specie controllate, fasi del ciclo biologico influenzate) ed in particolare alle proprietà profilattiche (ad esempio morbilità, mortalità, conteggio delle oocisti e quadro delle lesioni).
Devono essere fornite informazioni relative all'effetto sull'efficacia alimentare del mangime e sull'aumento di peso vivo.
I dati richiesti sull'efficacia comportano tre fasi di sperimentazione sugli animali bersaglio:
a) esperimenti controllati in batteria (infezioni singole e miste);
b) studi controllati all'interno di un box (condizioni simulate di impiego);
c) sperimentazioni controllate sul campo (condizioni reali di impiego).
Contemporaneamente e laddove ciò sia pertinente devono essere registrati, nell'ambito delle prove di efficacia, ulteriori dati per consentire una valutazione dell'interferenza con la crescita e con il tasso di conversione dei mangimi (volatili da ingrasso, pollastre da rimonta e conigli), degli effetti sulla fertilità delle uova e sulla loro schiusa (uccelli nidificanti).
2.2. Per altri additivi zootecnici
Devono essere fornite informazioni relative agli effetti sull'assunzione di mangime, sul peso corporeo, sull'assimilazione degli alimenti (preferibilmente riferita alla sostanza secca), sulla qualità e sulla resa del prodotto, nonché su qualsiasi altro parametro che abbia un'influenza positiva sull'animale, sull'ambiente, sul produttore o sul consumatore. Gli studi devono comprendere, laddove opportuno, un'indicazione del rapporto dose/risposta.
2.3. Condizioni sperimentali
Le sperimentazioni devono essere condotte perlomeno in due sedi diverse. I dati verranno riportati separatamente, con informazioni dettagliate sui controlli e su ciascun trattamento sperimentale. Per quanto riguarda i dati descrittivi generali, il protocollo della sperimentazione deve essere attentamente preparato nella forma seguente.
2.3.1. Mandria o gregge: ubicazione e dimensioni; condizioni di alimentazione e allevamento metodo di alimentazione; per le specie acquatiche dimensione e numero delle vasche o delle reti nell'allevamento e qualità dell'acqua.
2.3.2. Animali: specie (per specie acquatiche destinate al consumo umano esse vengono identificate utilizzando la denominazione non scientifica seguita tra parentesi dalla denominazione in latino o linneana), razza, età, sesso, procedura di identificazione, fase fisiologica e salute generale.
2.3.3. Numero dei test e dei gruppi di controllo, numero di animali in ciascun gruppo. Il numero dagli animali coinvolti nelle sperimentazioni deve consentire l'analisi statistica. Devono essere indicati i metodi di valutazione statistica utilizzati. Devono essere fornite almeno tre sperimentazioni confrontabili indipendenti con livello di probabilità pari a p < 0,05 per ciascuna categoria di animali interessata per dimostrare l'effetto indicato. Nel caso dei ruminanti potrebbe essere accettato un più basso livello di probabilità pari a p < 0,10. La relazione deve riguardare tutti gli animali o tutte le unità sperimentali coinvolti nelle sperimentazioni. Devono essere indicati i casi che non possono essere valutati a causa della mancanza o della perdita di dati e deve essere classificata la loro distribuzione all'interno dei gruppi di animali.
2.3.4. Diete: descrizione delle modalità di fabbricazione e della composizione quantitativa delle diete indicando gli ingredienti utilizzati ed i relativi principi nutritivi pertinenti (valori analitici) ed energia. Documentazione relativa all'assunzione di mangime.
2.3.5. Deve essere stabilita la concentrazione negli alimenti per animali del principio attivo (e se del caso delle sostanze utilizzate a fini comparativi) mediante un'analisi di controllo, che si avvalga di un appropriato metodo riconosciuto. Indicare i numeri di riferimento dei lotti.
2.3.6. Data e durata esatta delle prove. Data e natura degli esami eseguiti.
2.3.7. Studi di determinazione della dose: lo scopo di questi studi è spiegare la motivazione della scelta di una dose o di un intervallo di dosaggio dichiarati come pienamente efficaci. La determinazione della dose si baserà su un controllo (senza somministrazione di antibiotici, di fattori della crescita o di altre sostanze medicamentose) e su almeno tre livelli diversi da zero negli animali bersaglio.
2.3.8. Devono essere indicati i tempi e la prevalenza di eventuali conseguenze indesiderabili del trattamento in singoli o gruppi (devono essere fornite informazioni dettagliate del programma di osservazione utilizzato nello studio).
2.3.9. Tutti gli additivi studiati nelle condizioni di allevamento devono offrire valide prove scientifiche di sicurezza per l'utilizzatore, il consumatore, l'animale e l'ambiente. Quando un additivo non soddisfa i requisiti per la sicurezza del consumatore, qualsiasi studio deve essere progettato in modo di impedire l'immissione nella catena alimentare dei prodotti animali ottenuti dagli animali da esperimento.
III.3. Studi della qualità dei prodotti di origine animale.
I prodotti animali devono essere, a seconda dei casi, esaminati per le loro proprietà organolettiche, nutrizionali, igieniche e tecnologiche.
III.4. Studi relativi agli effetti sulle caratteristiche delle deiezioni animali.
Se l'additivo è destinato a modificare alcune caratteristiche delle deiezioni animali (ad esempio azoto, fosforo, odore, volume), sono necessari studi che dimostrino tali proprietà.
Capitolo IV: Studi relativi alla sicurezza d'impiego dell'additivo
Gli studi delineati in questo Capitolo sono destinati a consentire la valutazione di quanto segue:
sicurezza dell'impiego dell'additivo nella specie bersaglio;
qualsiasi rischio collegato alla selezione e/o al trasferimento della resistenza agli antibiotici nonché qualsiasi rischio di maggiore persistenza e/o riassorbimento degli enteropatogeni;
i rischi per il consumatore che potrebbero derivare dal consumo di prodotti alimentari contenenti residui dell'additivo o dei suoi metaboliti;
i rischi da inalazione o da contatto con altre mucose, con occhi o pelle per le persone che devono manipolare l'additivo in quanto tale o incorporato nelle premiscele o negli alimenti per animali;
i rischi di effetti negativi sull'ambiente dovuti all'additivo stesso od ai prodotti da esso derivati direttamente e/o per escrezione da parte degli animali.
Devono essere prese in considerazione le incompatibilità e/o le interazioni note tra l'additivo e i medicinali veterinari e/o i principali componenti della dieta, che concernono la specie interessata. Tali studi sono di norma tutti richiesti per ciascun additivo, salvo che non sia prevista una specifica esclusione o modifica dalla normativa vigente.
La presentazione di studi più limitati sarà di norma accettata nel caso in cui la proposta riguardi l'estensione dell'autorizzazione all'impiego ad una specie fisiologicamente e metabolicamente vicina ad una specie nella quale l'impiego dell'additivo sia già stato autorizzato. Questo ridotto complesso di dati deve dimostrare la sicurezza per la nuova specie e la mancanza di differenze significative per quanto concerne il destino metabolico dell'additivo ed i residui nei tessuti commestibili. L'MRL ed il periodo di sospensione proposti per la specie in questione devono essere motivati.
Per valutare i rischi per il consumatore e di conseguenza ai fini della determinazione degli MRL e del periodo di sospensione devono essere fornite le seguenti informazioni:
struttura chimica del principio attivo;
metabolismo nelle specie bersaglio oggetto della proposta;
natura dei residui in tali specie bersaglio;
studio della deplezione tissutale dei residui;
dati relativi agli effetti biologici del principio attivo e dei suoi metaboliti.
Può essere utile conoscere anche la biodisponibilità dei residui (liberi e legati) nel caso in cui vengano cioè prodotti molti metaboliti e non venga dimostrata la presenza di residui marcatori (cfr. paragrafo IV.1, sub 1.3.3).
Occorre inoltre conoscere la composizione e le proprietà fisicochimiche e biologiche dei principali escreti provenienti dall'additivo, al fine di definire l'ambito degli studi necessari per la valutazione del rischio di effetti sfavorevoli sull'ambiente o di persistenza nell'ambiente (cfr. paragrafo IV.5)
IV.1. Studi sulle specie bersaglio.
1.1. Test di tolleranza nelle specie/categorie di animali bersaglio
L'obiettivo è quello di determinare un margine di sicurezza (cioè un margine tra il dosaggio massimo proposto negli alimenti per animali e il livello minimo associato ad effetti indesiderati).
Un margine di sicurezza con un fattore non inferiore a 10 viene comunque considerato sufficiente ad escludere ulteriori prove. Tale test di tolleranza deve essere condono nelle specie/categorie di animali bersaglio preferibilmente durante tutta la durata del periodo produttivo anche se di norma sarebbe accettabile un periodo di prova di un mese. Ciò comporta almeno la valutazione dei segni clinici e di altri parametri al fine di valutare gli effetti sulla salute degli animali bersaglio. Deve essere compreso un gruppo di controllo negativo (cui non siano somministrati antibiotici, fattori della crescita o altre sostanze medicamentose). Sulla base del profilo tossicologico possono essere necessari ulteriori parametri. In questa Capitolo devono essere segnalati anche eventuali effetti indesiderati rilevati durante le prove di efficacia.
Qualora il prodotto sia destinato ad essere impiegato in animali utilizzabili a fini riproduttivi, devono essere condotti studi per individuare possibili danni alla funzione riproduttiva generale maschile o femminile oppure gli effetti nocivi sulla prole derivanti dalla somministrazione dell'additivo studiato.
1.2. Sicurezza microbiologica dell'additivo.
1.2.1. Tutti gli studi devono essere condotti con il più elevato dosaggio proposto.
1.2.2. Se il principio attivo presenta attività antimicrobica al livello della sua concentrazione nel mangime, occorrerebbe determinare, sulla base di procedure normalizzate, la concentrazione minima inibente (MIC) nei batteri patogeni e non patogeni, endogeni ed esogeni che interessano.
1.2.3. Test per determinare la capacità dell'additivo di:
indurre resistenza crociata agli antibiotici in questione;
selezionare nelle condizioni reali d'uso ceppi batterici resistenti nelle specie bersaglio; in tal caso ricerche sui meccanismi genetici per il trasferimento dei geni di resistenza.
1.2.4. Test per determinare l'effetto dell'additivo:
su una serie di organismi patogeni opportunisti presenti nell'apparato digerente (ad esempio enterobacteriacee, enterococchi e clostridia);
sul riassorbimento o sull'escrezione di microrganismi zoonosici, ad esempio Salmonella spp, Campylobacter spp.
1.2.5. Nel caso in cui il principio attivo presenti un'azione antimicrobica, devono essere forniti studi sul campo per il monitoraggio della resistenza batterica all'additivo.
1.3. Studi del metabolismo e dei residui
1.3.1. Tau studi intendono:
determinare le vie metaboliche del principio attivo quale base della relativa valutazione tossicologica;
individuare i residui e determinarne la cinetica nei tessuti e nei prodotti commestibili (latte, uova);
individuare gli escreti e ciò quale condizione preliminare per la valutazione del loro impatto sull'ambiente;
Talvolta, ad esempio nel caso degli additivi costituiti da prodotti di fermentazione, potrebbe essere necessario estendere gli studi ad altre sostanze aggiunte od ottenute durante il processo di fermentazione. Un esempio in tal senso è dato dal possesso di una tossicità significativa rispetto a quella dei componenti attivi dell'additivo.
1.3.2. Farmacocinetica
La pianificazione e il modello sperimentale degli studi devono tener conto delle caratteristiche anatomiche, fisiologiche (età, tipo, sesso), della categoria zootecnica e delle particolarità ambientali della popolazione bersaglio. Laddove opportuno, va considerata l'incidenza della microflora intestinale o ruminale, del circolo enteroepatico o della cecotrofia. La posologia sperimentata deve essere quella destinata ad essere utilizzata ed eventualmente un multiplo di taledose laddove ciò sia motivato. Il principio attivo (compresa la sostanza marcata) deve essere incorporato nell'alimento per animali salvo che non ci sia una motivazione per non farlo.
Sono necessari i seguenti studi:
bilancio metabolico e cinetica nel plasma/sangue dopo la somministrazione di una sola dose per valutare il tasso e l'entità dell'assorbimento, della distribuzione e dell'eliminazione (urine, feci, branchie, bile, aria espirata, latte o uova);
individuazione dei principali (>10%) metaboliti negli escreti, salvo qualora un metabolita secondario (<10%) sembri rappresentare una preoccupazione d'ordine tossicologico;
distribuzione dei materiali marcati nei tessuti e nei prodotti a seguito della somministrazione di una sola dose ad animali in cui sia già stato raggiunto l'equilibrio dello stato stazionario con un additivo non marcato.
Gli studi di cui ai punti 1.3.1 e 1.3.2 devono comprendere le tecniche con traccianti isotopici o metodi alternativi pertinenti.
1.3.3. Studio dei residui
identificazione di quei residui [composto progenitore, metaboliti, prodotti di degradazione, residui legati] che rappresentano oltre il 10% del residuo totale (salvo qualora un metabolita secondario sembri rappresentare una preoccupazione d'ordine tossicologico) nei tessuti e nei prodotti commestibili (latte, uova) in condizioni di equilibrio metabolico, ovvero dopo la somministrazione di diverse dosi della sostanza marcata; rapporto tra il residuo marcatore ed il totale dei residui;
studio cinetico dei residui nei tessuti (compresi latte e uova laddove opportuno) nel corso del periodo di deplezione dopo il raggiungimento dello stato stazionario e mediante l'utilizzo del livello più elevato del profilo metabolico proposto, identificazione del tessuto bersaglio e del residuo marcatore;
studio della deplezione del residuo marcatore dai tessuti bersaglio (compresi latte e uova laddove opportuno) dopo la sospensione dell'additivo successiva alla somministrazione ripetuta in base alle condizioni di impiego proposte e sufficiente per il raggiungimento dello stato stazionario, in modo da fissare un periodo di sospensione sulla base dell'MRL stabilito;
il periodo di sospensione dell'additivo non deve avere durata inferiore al tempo necessario affinché la concentrazione del residuo marcatore individuato nel tessuto bersaglio scenda al di sotto del valore MRL (limite di affidabilità del 95%). Devono essere presi in considerazione come requisito minimo punti temporali tra loro distanziati, scelti in modo appropriato in ragione della fase di deplezione del principio attivo e dei suoi metaboliti, nonché almeno quattro animali per punto temporale in base alla specie (dimensioni, variabilità genetica)
IV.2. Studi su animali da laboratorio.
Tali studi devono essere effettuati sul principio attivo utilizzando metodi di prova normalizzati internazionalmente riconosciuti come quelli descritti nelle OECD Guidelines for methodological details (linee direttrici dell'OCSE sui dettagli metodologici) e sulla base dei principi delle buone prassi di laboratorio (BPL). Possono essere necessari ulteriori studi su determinati metaboliti prodotti dalla specie bersaglio qualora essi non si sviluppino in misura significativa nelle specie impiegate nei test di laboratorio. Quando sono disponibili dati relativi all'uomo, può essere necessario prendere in considerazione anche questo aspetto nel momento in cui si decida quali altri studi eseguire.
2.1. Tossicità acuta
Studi della tossicità orale acuta devono essere condotti almeno in due specie di mammiferi. Una specie di laboratorio può, laddove opportuno, essere sostituita da una specie bersaglio. Non occorrerà individuare una DL50 precisa; di norma è sufficiente una determinazione approssimativa della dose letale minima. Al fine di ridurre il numero e le sofferenze degli animali trattati, il dosaggio massimo non deve superare 2000 mg/kg pc e si raccomandano metodi alternativi (saggio limite, metodo a dose fissa, metodo della classe di tossicità acuta).
I rischi per i lavoratori devono essere valutati in una serie di studi nei quali venga utilizzato il prodotto (principio attivo ed eccipiente nella forma in cui il prodotto sarà disponibile in commercio). Devono essere condotti studi sul potere di irritazione cutanea e nel caso in cui risultino positivi occorrerebbe valutare il potere di irritazione delle membrane mucose (ad esempio occhi). Devono inoltre essere valutati il rischio allergenico e di sensibilizzazione della cute. Devono essere eseguiti studi sull'inalazione acuta nel caso in cui sia probabile che il prodotto dia origine ad una polvere o ad un vapore inalabili.
2.2. Studi di genotossicità, compresa la mutagenicità
Per identificare i principi attivi ed eventualmente i loro metaboliti e prodotti di degradazione con proprietà mutagene e genotossiche, deve essere eseguita una serie di almeno tre prove combinate di genotossicità. La batteria dei test deve di norma comprendere prove nei sistemi procariota ed eucariota, compresi sistemi di saggio in vitro e in vivo nei mammiferi. Laddove opportuno tale test devono essere condotti con e senza attivazione metabolica nei mammiferi.
La scelta dei test deve essere motivata sulla base della loro affidabilità nella valutazione degli effetti genotossici sui diversi loci genetici a livello genico, cromosomico e genomico. Possono essere opportune ulteriori prove in base all'esito dei test eseguiti e alla luce del profilo complessivo di tossicità della sostanza, nonché dell'uso che se ne intende fare. Le prove devono essere eseguite conformemente a procedure convalidate consolidate ed aggiornate. Quando il bersaglio del test è il midollo osseo, in presenza di un risultato negativo occorre fornire la prova dell'esposizione delle cellule alla sostanza in esame.
2.3. Studi della tossicità orale subcronica (novanta giorni)
Le prove devono durare almeno novanta giorni. Nel caso di additivi destinati ad essere utilizzati in specie animali da cui si ottengono prodotti alimentari, gli studi devono essere condotti su due specie animali, una delle quali deve essere una specie di non roditori, che potrebbe a sua volta essere la specie bersaglio. Nel caso di additivi destinati ad animali per i quali non è previsto il consumo da parte dell'uomo, sono sufficienti studi sulla specie bersaglio: al fine di ottenere la risposta alla dose, il principio attivo deve essere somministrato almeno a tre livelli oltre che al gruppo di controllo. La dose massima deve di norma produrre segni di effetti nocivi. Il dosaggio minimo non deve produrre alcun segno di tossicità.
2.4. Studi di tossicità orale cronica (compresi gli studi di cancerogenicita)
Uno studio della tossicità cronica, comprendente eventualmente un esame della cancerogenicità, deve essere condotto in almeno una specie di roditori.
Gli studi di cancerogenicità possono non essere necessari nel caso in cui il principio attivo ed i suoi metaboliti:
forniscano risultati costantemente negativi in una serie adeguata di test di genotossicità;
non siano strutturalmente correlati a noti agenti cancerogeni;
non producano effetti che facciano ipotizzare un rischio di (pre)neoplasia nelle prove di tossicità cronica.
2.5. Studi di tossicità riproduttiva, inclusa la teratogenicità
2.5.1. Studi di tossicità riproduttiva su due generazioni
Devono essere condotti studi relativi alla funzione riproduttiva, che devono estendersi su almeno due generazioni in linea diretta (F1, F2) e devono essere associati ad uno studio di teratogenicità. La sostanza in esame viene somministrata ai maschi e alle femmine a partire da un certo momento prima dell'accoppiamento e la somministrazione deve proseguire fino allo svezzamento della generazione F2.
Devono essere analizzati e segnalati tutti i dati pertinenti relativi alla fertilità, alla gestazione, al parto, al comportamento della madre, all'allattamento, all'accrescimento e allo sviluppo della generazione F1 dalla fecondazione fino alla maturità e allo sviluppo della generazione F2 fino allo svezzamento.
2.5.2. Studio di teratogenicità
Lo studio di teratogenicità riguarda la tossicità embrio-fetale. Deve essere condotto in almeno due specie.
2.6. Studi del metabolismo e dell'eliminazione
Devono essere condotti studi dell'assorbimento, della distribuzione nei liquidi e nei tessuti organici e delle vie di eliminazione. Deve essere condotto uno studio metabolico che comprenda il bilancio metabolico e l'identificazione dei principali metaboliti nelle urine e nelle feci in animali di entrambi i sessi ed appartenenti agli stessi ceppi di quelli utilizzati negli studi tossicologici. Deve essere somministrata un'unica dose della molecola marcata (cfr. 4.1.3), una volta raggiunto lo stato stazionario d'equilibrio utilizzando il composto non marcato in un dosaggio simile al dosaggio più elevato proposto per l'utilizzo nell'animale bersaglio.
2.7. Biodisponibilità dei residui
La valutazione del rischio per i consumatori connesso a determinati residui contenuti nei prodotti di origine animale, ovvero relativo ai residui legati, può tener conto di un ulteriore fattore di sicurezza basato sulla determinazione della loro biodisponibilità mediante l'utilizzo di idonei animali di laboratorio e appropriati metodi riconosciuti.
2.8. Altri studi tossicologici e farmacologici specifici
Laddove esistano motivi di preoccupazione, devono essere condotti ulteriori studi in grado di fornire ulteriori informazioni utili per la valutazione della sicurezza dei principio attivo e dei suoi residui.
2.9. Determinazione di una dose senza effetto osservato (NOEL)
Al fine di individuare un NOEL espresso come mg/kg di peso corporeo al giorno, devono essere presi in considerazione tutti i risultati sopraindicati ed i dati pubblicati pertinenti (comprese eventuali opportune informazioni sugli effetti del principio attivo sull'uomo), nonché, laddove ciò sia opportuno, le informazioni su strutture chimiche strettamente correlate. Deve essere prescelto il NOEL più basso.
Ciononostante il NOEL da utilizzare per il calcolo della dose giornaliera accettabile (DGA) deve, a seconda dei casi, essere scelto sulla base degli effetti tossicologici o farmacologici. Per quanto concerne alcuni additivi, ad esempio gli antibatterici, può essere meglio fissare la DGA sulla base degli effetti sulla microflora dell'intestino umano. In mancanza di metodi per descrivere la flora intestinale che siano convalidati ed accettati a livello internazionale, possono risultare più opportuni gli effetti su ceppi batterici selezionati e sensibili dell'intestino umano.
IV.3. Valutazione della sicurezza per il consumatore umano.
3.1. Proposta di una dose giornaliera accettabile (DGA) per l'additivo.
Laddove opportuno, deve essere proposta una DGA.
La DGA (espressa come milligrammi di additivo o materiale ad esso connesso per persona al giorno) si ottiene dividendo il NOEL (dose senza effetto osservato) per un opportuno fattore di sicurezza e moltiplicando per il peso corporeo umano medio (pc) di 60 kg. Il NOEL espresso quindi come milligrammi per chilogrammo di peso corporeo può essere scelto utilizzando i risultati tossicologici o farmacologici. In alcuni casi può essere maggiormente pertinente una DGA basata sulle proprietà microbiologiche degli additivi. La scelta dipende dalla proprietà maggiormente pertinente in termini di rischio per la salute del consumatore.
Il fattore di sicurezza utilizzato nel determinare la DGA di un determinato additivo deve essere prescelto tenendo presente:
la natura dell'effetto biologico utilizzato per individuare il NOEL;
la pertinenza di tale effetto rispetto all'uomo e la reversibilità dell'effetto medesimo;
la gamma e la qualità dei dati utilizzati per individuare il NOEL;
eventuali effetti noti dei componenti del residuo.
Nel calcolo della DGA viene di solito utilizzato un fattore di sicurezza pari almeno a 100 (cioè un fattore 10 per tener conto della possibile variazione tra le specie ed un ulteriore fattore 10 per tener conto di possibili risposte diverse tra esseri umani). Qualora siano disponibili i dati sul principio attivo relativi all'uomo può essere accettabile un più basso fattore di sicurezza.
3.2. Proposta dei limiti massimi di residui (MRL) dell'additivo
Il calcolo dell'MRL si fonda sull'ipotesi che l'unica fonte di possibile esposizione dell'uomo sia rappresentata dall'assunzione di tessuto commestibile, latte ed ovoprodotti. Altrimenti occorre tener conto delle altre fonti.
Numerose sostanze di questo tipo sono state utilizzate quali additivi nei mangimi e per altre applicazioni. In tali casi gli MRL devono essere gli stessi. In alcuni casi, sulla base di rigorose valutazioni scientifiche, possono anche essere calcolati MRL diversi per ciascun utilizzo; questo accade quando la via di somministrazione, la quantità, la frequenza e la durata della somministrazione sono sufficientemente diverse da quelle previste per l'utilizzo quale mangime ed esistono prove in grado di indicare che la cinetica e/o il metabolismo potrebbero dar luogo a profili diversi dei residui. In tali casi si prevede l'applicazione dell'MRL più rigoroso.
Per fissare un MRL, occorre definire la natura chimica della sostanza farmaco-correlata da utlizzare ai fine di specificare i livelli dei residui nel tessuti. Ciò è noto come residuo marcatore. Questo componente di residuo non deve necessariamente essere il residuo di interesse tossicologico, ma deve essere prescelto quale indicatore idoneo a rappresentare il totale del residuo significativo. Nel corso degli studi relativi alla deplezione devono essere determinati, ad ogni punto temporale, i rapporti residuo marcatore/residui totali in relazione alla DGA (ovvero rapporto residuo marcatore/residui radioattivi totali, residuo marcatore/tutti i residui biologicamente attivi). Questo rapporto deve, in particolare, essere noto per il punto temporale prescelto per determinare gli MRL. Occorre inoltre disporre di un metodo analitico idoneo per questo residuo marcatore in modo da garantire il rispetto dell'MRL.
Nel determinare gli MRL (espressi in g/Kg di residuo marcatore per Kg di tessuto o prodotto commestibile umido) sulla base di una data DGA, devono essere applicati i seguenti valori per quanto concerne il consumo giornaliero di alimenti da parte dell'uomo:
|
|
Mammiferi |
Uccelli |
Pesci |
|
Muscolo ........................ |
300 g |
300 g |
300 g |
|
Fegato .......................... |
100 g |
100 g |
|
|
Rene ............................. |
50 g |
10 g |
|
|
Grasso .......................... |
50 g (**) |
90 g (***) |
|
|
+ latte ............................ |
1500 g |
|
|
|
+ uova ............................ |
|
100 g |
|
(*)Muscolo e pelle nelle proporzioni naturali;
(**) Per i suini 50 grammi di grasso e pelle nelle proporzioni naturali;
(***) Grasso e pelle nelle proporzioni naturali.
I singoli MRL nei diversi tessuti devono riflettere la cinetica della deplezione dei residui nei tessuti delle specie animali nei quali si intende utilizzare la sostanza in questione. Occorre un metodo analitico con un limite di quantificazione inferiore all'MRL (cfr. Capitolo II, punto 5.3).
Qualora la sostanza possa dare origine ad un residuo nei tessuti e nei prodotti, occorrebbe proporre MRL tali che il totale del residuo tossicologicamente (o microbiologicamente) significativo ingerito giornalmente sia inferiore alla DGA (cfr. tabella sopra).
L'MRL deve essere fissato solo dopo aver esaminato e tenuto conto di ogni altra possibile fonte di esposizione del consumatore ai componenti dei residui.
Nel caso di alcuni additivi, si potrebbero formare nel latte, nelle uova o nella carne residui al di sotto degli MRL in grado comunque di interferire con la qualità dei prodotti aumentati in determinati processi di trasformazione alimentare, quale ad esempio l'utilizzo del latte nella fabbricazione del formaggio. Nel caso di tali additivi, oltre a fissare gli MRL, può risultare opportuno prendere in considerazione un residuo massimo compatibile con la trasformazione (alimentare).
In alcuni casi non sarà necessario un MRL, ad esempio quando:
non vi è alcuna biodisponibilità dei residui e non vi è alcun effetto nocivo sull'intestino umano, compresa la relativa microflora;
si assiste ad una completa degradazione in sostanze nutritive o innocue nelle specie bersaglio;
la DGA è "non specificata" a causa di una ridotta tossicità nelle prove sugli animali;
l'utilizzo è limitato esclusivamente ad alimenti per gli animali da compagnia;
una sostanza è approvata anche quale additivo alimentare; in tal caso un MRL non è di norma richiesto qualora il residuo marcatore sia principalmente rappresentato dalla sostanza madre e costituisca solo una percentuale esigua della DGA dell'additivo alimentare.
3.3. Proposta del periodo di sospensione relativo all'additivo.
Il periodo di sospensione verrà fissato sulla base degli MRL. Il periodo di sospensione comprende il periodo successivo all'interruzione della somministrazione dell'additivo nella formulazione proposta;
periodo necessario per consentire un abbassamento dei livelli dei residui al di sotto degli MRL (limite di affidabilità del 95%).
Per determinare un periodo di sospensione, può essere individuato un determinato tessuto commestibile sostitutivo di altri, al quale viene dato il nome di tessuto bersaglio.
IV.4. Valutazione della sicurezza dei lavoratori.
I lavoratori possono essere esposti per inalazione oppure per via topica durante la fabbricazione oppure la manipolazione o l'utilizzo dell'additivo, come ad esempio nel caso della manodopera occupata in agricoltura che rischia di essere esposta durante la manipolazione o la miscelazione dell'additivo. Devono essere fornite ulteriori informazioni sulle modalità di manipolazione delle sostanze. Deve essere compresa una valutazione del rischio per i lavoratori. Un'importante fonte di informazioni per la valutazione dei rischi per i lavoratori derivanti dall'esposizione all'additivo per inalazione o per via topica è costituita dalla conoscenza dell'impianto di produzione. Particolare preoccupazione destano gli additivi/gli alimenti trattati con additivi e/o gli escreti animali che sono costituiti da polveri secche o che possono dar origine a polveri secche e gli additivi dei mangimi che possono presentare rischi allergenici.
4.1. Valutazione del rischio tossicologico per la sicurezza dei lavoratori
4.1.1. Effetti sul sistema respiratorio
Devono essere fornite prove che dimostrino che le polveri in sospensione non rappresenteranno un pericolo per la salute dei lavoratori. Tra tali prove devono figurare, laddove necessario, test per inalazione negli animali da laboratorio, dati epidemiologici pubblicati e/o i dati del richiedente relativi al proprio impianto e/o i test di irritazione cutanea e di sensibilizzazione respiratoria.
4.1.2. Effetti sugli occhi e sulla cute
Laddove disponibili, devono essere fornite prove dirette, relative a situazioni note in cui sia coinvolto l'uomo, dell'assenza di potere di irritazione e/o di sensibilizzazione. Queste prove devono essere integrate dai risultati di prove convalidate su animali concernenti l'irritazione cutanea ed oculare ed il rischio di sensibilizzazione associati all'utilizzo dell'additivo in questione.
4.1.3. Tossicità sistemica
l dati sulla tossicità elaborati per soddisfare i requisiti di sicurezza (comprese le prove di tossicità a dosi ripetute, mutagenicità, cancerogenicita) devono essere utilizzati per valutare altri aspetti della sicurezza dei lavoratori. Nel far ciò, occorre ricordare che le vie più probabili di esposizione sono rappresentate dalla contaminazione della cute e/o dall'inalazione dell'additivo.
4.2. Valutazione dell'esposizione
Devono essere fornite informazioni su come l'utilizzo dell'additivo potrebbe dar luogo ad ogni forma di esposizione: per inalazione, per via cutanea o per ingestione. Tali informazioni devono comprendere una valutazione quantitativa, laddove essa sia disponibile, riguardante ad esempio la tipica concentrazione atmosferica, la contaminazione cutanea o l'ingestione. Qualora non siano disponibili informazioni quantitative, devono essere fornite informazioni sufficienti che consentano di valutare adeguatamente l'esposizione.
4.3. Misure per controllare l'esposizione
Sulla base delle informazioni relative alla valutazione tossicologica e alla valutazione dell'esposizione deve essere tratta una conclusione sui rischi per la salute degli utenti (rischio sistemico, tossicità, potere di irritazione e sensibilizzazione) associati a misure ragionevoli per il controllo dell'esposizione. Qualora il rischio sia inaccettabile, devono essere adottate misure precauzionali per controllare od eliminare l'esposizione. Soluzioni preferibili sono rappresentate da una nuova formulazione del prodotto o dalla modifica delle procedure di produzione, utilizzo e/o smaltimento dell'additivo. L'utilizzo di dispositivi di protezione individuale deve essere considerato come l'ultima possibilità di protezione da eventuali rischi residui, una volta le misure di controllo.
IV.5. Valutazione del rischio ambientale.
E' importante considerare l'impatto ambientale degli additivi degli alimenti per animali in quanto la loro somministrazione interessa di solito un lungo arco di tempo (anche l'intero arco della vita), in quanto possono essere coinvolti grossi gruppi di animali e molti additivi sono scarsamente assorbiti e pertanto vengono in larga parte eliminati in forma non degradata. Ciononostante in molti casi può risultare ridotta la necessità di una valutazione ambientale. Non è opportuno fissare norme rigide in questa guida generale. Allo scopo di concorrere alla determinazione dell'impatto ambientale di un additivo degli alimenti per animali, deve essere seguito un approccio graduale (cfr. l'albero decisionale), che prevede che nella prima fase vengano individuati chiaramente gli additivi per i quali non è necessaria alcuna ulteriore sperimentazione. Per altri additivi occorre una seconda fase di studi (fase II A) in modo da disporre di ulteriori informazioni, in base alle quali potrebbero risultare necessari ulteriori studi (fase II B). Gli eventuali studi devono essere condotti a norma del decreto ministeriale 10 aprile 2000, recante recepimento direttiva 67/548/CEE
5.1. Valutazione - Fase I
La finalità della fase I della valutazione è quella di determinare la probabilità di un effetto significativo dell'additivo o dei suoi metaboliti sull'ambiente, principalmente sulla base di dati già raccolti per altri scopi.
Una dispensa dall'obbligo della fase II della valutazione può essere accordata sulla base di uno dei due criteri seguenti:
a) la natura chimica e l'effetto biologico dell'additivo nonché il suo utilizzo indicano che il suo impatto sarà trascurabile: ciò nel caso in cui l'additivo e/o i suoi principali metaboliti (costitutivi di oltre il 20% dei residui totali negli escreti) siano:
sostanze fisiologiche/naturali (ad esempio una vitamina o un minerale) che non altereranno la concentrazione ambientale, salvo che non ci siano evidenti motivi di preoccupazione (ad esempio il rame),
additivi destinati ad essere utilizzati negli animali da compagnia (esclusi i cavalli);
b) la peggiore concentrazione ambientale prevista (PEC) è troppo modesta per destare preoccupazione.
E' probabile che la peggiore PEC nel suolo sia conseguente allo spandimento sul terreno del mangime organico prodotto durante l'eliminazione dei principali componenti del residuo (l'additivo e/o i suoi principali metaboliti). La PEC deve essere valutata per ciascun componente principale del residuo nel mangime organico come pure per ogni comparto interessato. Per quanto concerne il comparto ambientale terrestre, se la PEC non supera 100 g/kg per l'insieme dei principali componenti del residuo oppure (qualora tali dati siano disponibili) se i principali componenti del residuo nel mangime organico vengono rapidamente degradati (tempo di degradazione DT 50 <30 giorni) in componenti naturali o a concentrazioni inferiori a 100 g/kg, oppure se la PEC nel suolo (ad una profondità di 5 cm) 6 inferiore a 10 g/kg, allora non occorre alcuna ulteriore valutazione. La peggiore PEC nell'acqua può essere la conseguenza del passaggio diretto in corpi idrici (a causa di fuoriuscite) di alimenti o escreti contenenti l'additivo e i suoi metaboliti oppure della lisciviazione nelle falde freatiche di materiali contenuti negli escreti o nel suolo. Qualora sulla base di una stima affidabile la PEC relativa alla contaminazione dei corpi idrici o delle acque freatiche si collochi al di sotto di 0,1 g per litro, non occorre alcuna fase II A di valutazione dell'impatto ambientale dell'additivo sul comparto delle acque.
Se il richiedente non è in grado di dimostrare che l'additivo proposto rientra in una delle categorie soggette all'esenzione di un'ulteriore valutazione oppure qualora l'additivo venga rilasciato direttamente nell'ambiente (ad esempio acquacoltura), sarà di norma necessaria la fase II della valutazione.
RISCHIO AMBIENTALE DERIVANTE DAGLI ADDITIVI DEGLI ALIMENTI PER ANIMALI
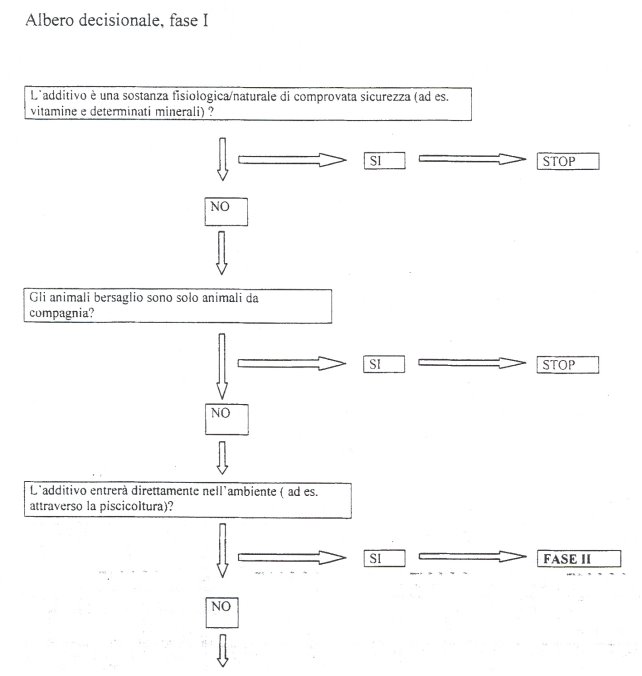
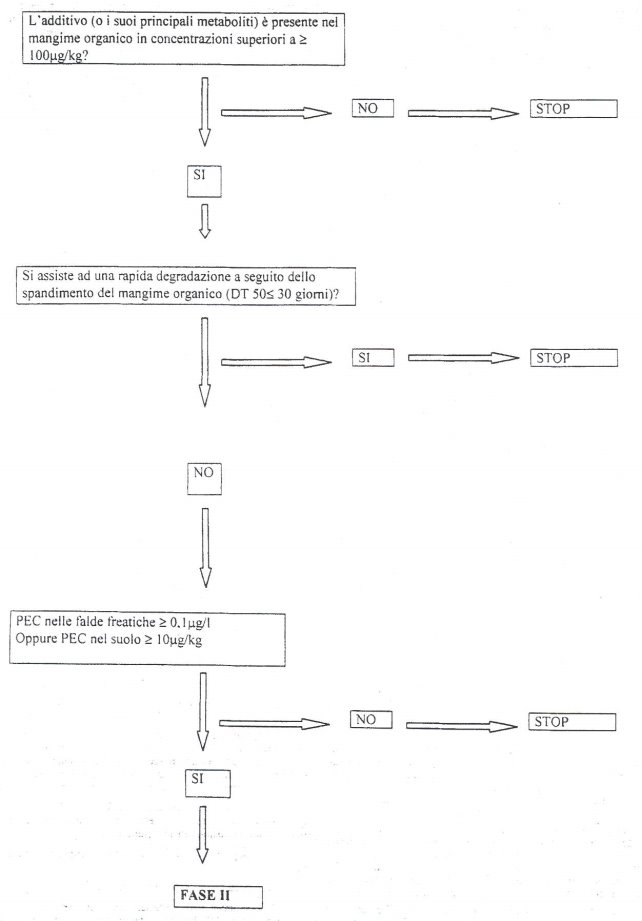
5.2. Fase II della valutazione
La fase due della valutazione consta di due fasi: fase II A e fase II B.
Devono essere valutati il rischio di bioaccumulazione dell'additivo e dei suoi principali metaboliti nonché la sua influenza sul margine di sicurezza previsto. La bioaccumulazione non viene ritenuta potenzialmente significativa qualora ad esempio il Kow (coefficiente di distribuzione) sia inferiore a 3. Di norma occorreranno opportuni studi della fase II B se non sarà possibile determinare tali margini di sicurezza.
5.2.1. Fase II A
La fase II A della valutazione intende valutare il rischio ambientale mediante:
un calcolo più accurato delle PEC;
la determinazione del rapporto tra esposizione, livelli di additivo e/o principali metaboliti e gli effetti indesiderati a breve termine nelle relative specie animali e vegetali sostitutive a livello dei comparti ambientali interessati;
l'utilizzo di tali risultati per determinare i valori delle prevedibili concentrazioni prive di effetti (PNEC).
Nella determinazione del rischio si raccomanda la seguente procedura di tipo sequenziale.
a) Deve essere calcolata una PEC più precisa per ciascun comparto ambientale interessato, qualora ciò non sia stato compiutamente effettuato nella fase I. Nel determinare la PEC si deve tener conto di quanto segue:
la concentrazione dell'additivo e/o dei suoi principali metaboliti nel mangime organico a seguito della somministrazione dell'additivo stesso agli animali nel dosaggio proposto. In tale calcolo devono essere presi in considerazione il volume degli escreti ed i tassi di dosaggio;
la potenziale diluizione degli escreti collegati all'additivo, a causa delle normali pratiche di trasformazione e conservazione del mangime organico prima del suo spargimento sul terreno;
assorbimento/desorbimento dell'additivo e dei suoi metaboliti nel suolo, persistenza dei residui nel suolo (DT50 e DT90); sedimento nel caso dell'acquacoltura;
altri fattori quali la fotolisi, l'idrolisi, l'evaporazione, la degradazione nel suolo o nel sedimenti dell'acqua, diluizione mediante la lavorazione del terreno, ecc.
Ai fini della valutazione del rischio di livello II A, si deve utilizzare il valore più alto di PEC ottenuto per ciascun comparto ambientale interessato attraverso questi calcoli.
Può essere necessaria un'ulteriore valutazione di livello II B qualora venga prevista un'elevata persistenza nel suolo (DT90 > 1 anno) a concentrazioni superiori a 10 g/kg di suolo allo stato stazionario.
b) Successivamente devono essere determinati i livelli responsabili di gravi effetti indesiderati a breve termine in relazione ai vari livelli trofici dei comparti ambientali interessati (suolo, acqua). Tali prove devono essere condotte secondo le linee direttrici dell'OCSE oppure secondo analoghe linee direttrici già consolidate. Prove idonee per l'ambiente terrestre comprendono: tossicità per i lombrichi (concentrazione letale nel 50% dei casi, CL50), fitotossicità (concentrazione efficace nel 50% dei casi, CE50) nelle piante terrestri, effetti sui microrganismi del terreno (ad esempio CE50 per gli effetti sulla metanogenesi e la fissazione dell'azoto). Per quanto riguarda l'ambiente acquatico, studio della CL50 a 96 ore nel pesce, studio della CE50 a 48 ore nella Daphnia magna, studio della CL50 nelle alghe ed uno studio di tossicità per gli organismi dei sedimenti.
c) Deve essere calcolato il valore della PNEC per ciascun comparto interessato. Ciò viene di norma ottenuto prendendo il più basso valore osservato (cioè il risultato relativo alla specie più sensibile) di un effetto sfavorevole riscontrato nelle prove di ecotossicità sopraindicate, dividendo poi per un fattore di sicurezza pari almeno a 100 a seconda dell'indicatore utilizzato e del numero delle specie studiate.
d) Occorrerebbe confrontare i valori PEC e PNEC calcolati. Il rapporto accettabile tra PEC e PNEC dipenderà dalla natura del risultato della prova utilizzato per determinare la PNEC. Di solito sarà compreso tra 1 e 0,1. Qualora vengano riscontrati rapporti significativamente inferiori a questi, è improbabile che siano necessarie ulteriori prove ecotossicologiche salvo qualora sia prevista una bioaccumulazione. Rapporti più elevati imporranno invece alcune prove della fase II B.
5.2.2. Fase II B (studi tossicologici maggiormente dettagliati)
Nel caso degli additivi per i quali, al termine della fase II A della valutazione, permangano dubbi circa l'impatto ambientale occorrono studi maggiormente approfonditi relativi agli effetti sulle specie biologiche dei comparti ambientali per i quali possibili preoccupazioni sono emerse a seguito degli studi della fase II A. In tale situazione occorrono ulteriori prove per determinare gli effetti cronici e quelli più specifici nelle opportune specie animali, vegetali o microbiche. E' possibile che la fase II A della valutazione abbia comportato una sopravvalutazione della PEC. Per dimostrarlo può essere necessario effettuare misurazioni delle concentrazioni ambientali e della persistenza dell'additivo e/o dei suoi principali metaboliti nelle situazioni reali di utilizzo.
Numerose pubblicazioni, ad esempio le linee direttrici dell'OCSE descrivono ulteriori opportune prove di ecotossicità. Può essere necessario esaminare tre categorie di specie ambientali: animali, piante e microrganismi. Tali prove devono essere attentamente selezionate in modo da garantire che esse siano adatte rispetto alla situazione dell'eventuale rilascio e dispersione dell'additivo e/o dei suoi metaboliti nell'ambiente.
La valutazione dell'impatto sul comparto terrestre può comprendere:
uno studio degli effetti sub letali sui lombrichi, ulteriori studi dell'impatto sulla microflora del terreno, prove di fitotossicità su una serie di specie vegetali economicamente importanti, studi sugli invertebrati presenti nei pascoli, inclusi gli insetti, e sugli uccelli selvatici.
N.B. La valutazione della tossicità sui mammiferi può non essere necessaria in quanto questo aspetto deve essere preferibilmente affrontato mediante le prove di tossicità sui mammiferi condotte per determinare la DGA.
La valutazione dell'impatto sul comparto acquatico può comprendere:
prove di tossicità cronica sugli organismi acquatici più sensibili identificati nella fase II A della valutazione, ad esempio prove sui pesci nelle prime fasi della vita, il saggio sulla riproduzione della Daphnia, il test nelle 72 ore sulle alghe ed uno studio di bioaccumulazione.
Quando non può essere identificato un idoneo margine di sicurezza tra i valori della PEC e della PNEC, devono essere indicate misure efficaci che consentano di attenuare l'impatto sull'ambiente.
Capitolo V: Modello di monografia
V.1. Identità dell'additivo.
1.1. Denominazioni commerciali proposte.
1.2. Tipo di additivo in base alla sua funzione principale. Devono essere precisati eventuali altri impieghi del principio attivo.
1.3. Composizione qualitativa e quantitativa (principio attivo, altri componenti, impurità, variazione tra le partite). Se il principio attivo è costituito da una miscela di vari componenti attivi ciascuno dei quali chiaramente definibile, occorre descrivere separatamente i principali componenti e indicare le loro proporzioni nella miscela.
1.4. Stato fisico, distribuzione delle particelle in base alla loro dimensione, forma delle particelle, densità, peso specifico apparente; per i liquidi: viscosità, tensione superficiale.
1.5. Procedimento di fabbricazione, compresi eventuali trattamenti specifici.
V.2. Specifiche relative al principio attivo.
2.1. Denominazione generica, denominazione chimica secondo la nomenclatura UICPA. altre denominazioni generiche internazionali e abbreviazioni. Numero CAS (Chemical Abstracts Service Number).
2.2. Formula di struttura, formula molecolare e peso molecolare. Composizione qualitativa e quantitativa dei principali componenti, origine microbica (denominazione e sede della raccolta delle colture presso cui il ceppo è depositato) se il principio attivo è un prodotto di fermentazione.
2.3. Purezza
Composizione qualitativa e quantitativa dei principi attivi e delle impurità e delle sostanze tossiche ad essi associate, conferma dell'assenza di organismi di produzione.
2.4. Proprietà pertinenti
Proprietà fisiche delle sostanze chimicamente specificate: costante di dissociazione, pKa, proprietà elettrostatiche, punto di fusione, punto di ebollizione, densità, pressione di vapore, solubilità in acqua e in solventi organici, Kow e Koc, spettri di massa e di assorbimento, dati RMN, possibili isomeri ed ogni altra opportuna proprietà fisica.
V.3. Proprietà fisicochimiche, tecnologiche e biologiche dell'additivo.
3.1. Stabilità dell'additivo rispetto alle condizioni ambientali, quali luce, temperatura, pH, umidità, ossigeno. Proposta di una durata di conservazione.
3.2. Stabilità durante la preparazione delle premiscele e degli alimenti per animali, in particolare stabilità rispetto alle previste condizioni di lavorazione (calore, umidità, pressione/sollecitazione di taglio e tempo). Possibili prodotti di degradazione o decomposizione.
3.3. Stabilità durante la conservazione delle premiscele e degli alimenti per animali lavorati, in presenza di determinate condizioni. Proposta di una durata di conservazione.
3.4. Altre proprietà fisiochimiche, tecnologiche o biologiche, quali la possibilità di dispersione in presenza di condizioni favorevoli, in modo da ottenere e conservare miscele omogenee nelle premiscele e negli alimenti, proprietà antipolvere ed antistatiche, possibilità di dispersione nei liquidi.
V.4. Metodi di controllo.
4.1. Descrizione dei metodi utilizzati per la determinazione dei criteri elencati ai punti 1.3, 1.4, 2.3, 2.4, 3.1, 3.2, 3.3 e 3.4 del capitolo II.
4.2. Descrizione dei metodi analitici qualitativi e quantitativi per la determinazione dei residuo marcatore del principio attivo nei tessuti bersaglio e nei prodotti di origine animale.
4.3. Se tali metodi sono stati pubblicati nella letteratura, è sufficiente allegarne copia con indicazione dei riferimenti bibliografici.
4.4. Informazioni sulle condizioni di conservazione ottimali dei campioni di riferimento.
V.5. Proprietà biologiche dell'additivo.
5.1. Informazioni dettagliate sugli effetti profilattici dei coccidiostatici e di altre sostanze medicamentose (ad esempio morbilità, mortalità, conteggio delle oocisti e quadro delle lesioni).
5.2. Per gli additivi zootecnici diversi da quelli elencati al punto 5.5.1 devono essere fornite informazioni dettagliate relative agii effetti sull'assunzione di mangime, sul peso corporeo, sull'assimilazione degli alimenti, sulla qualità e sulla resa del prodotto, nonché su qualsiasi altro parametro che abbia un'influenza positiva sull'animale, sull'ambiente, sul produttore o sul consumatore.
5.3. Per gli additivi tecnologici, effetti tecnologici pertinenti.
5.4. Eventuali effetti indesiderati, controindicazioni o avvertenze per l'animale bersaglio, il consumatore, l'ambiente, comprese le interazioni biologiche, con l'indicazione delle relative giustificazioni. Devono essere precisati laddove possibile la DGA o gli MRL eventualmente fissati per altri utilizzi.
Per questi prodotti deve, inoltre, essere fornita una dettagliata informazione sugli eventuali residui quantitativi e qualitativi nei tessuti bersaglio rilevati nei prodotti di origine animale successivamente all'impiego previsto dell'additivo. Laddove opportuno, devono essere indicati la DGA, gli MRL previsti e il periodo di sospensione.
5.6. Caratteristiche atte ad identificare l'additivo
5.7. Devono essere riportate con puntualità le condizioni di impiego
V.6. Data.
Capitolo VI: Struttura della scheda segnaletica
VI.1. Identità dell'additivo.
1.1. Tipo di additivo
1.2. Stato fisico
1.3. Composizione qualitativa e quantitativa
1.4. Metodo di analisi dell'additivo e dei residui
1.5. Numero di registrazione comunitario (numero CE)
1.6. Imballaggio
VI.2. Specifiche relative al principio attivo.
2.1. Denominazione generica, denominazione chimica, numero CAS
Denominazione generica
Denominazione chimica (UICPA)
Numero CAS
2.2. Formula empirica
VI.3. Proprietà fisicochimiche, tecnologiche e biologiche dell'additivo.
3.1. Stabilità dell'additivo
3.2. Stabilità durante la preparazione delle premiscele e degli alimenti per animali
3.3. Stabilità durante la conservazione delle premiscele e degli alimenti per animali
3.4. Altre proprietà
VI.4. Condizioni di impiego.
4.1. Specie o categoria di animali, età massima se indicata
4.2. Contenuto minimo e massimo negli alimenti per animali
4.3. Controindicazioni, interazioni
4.4. Avvertenze
VI.4. Responsabile dell'immissione in circolazione.
5.1. Nome
5.2. Indirizzo
5.3. Numero di registrazione
VI.6. Fabbricante.
6.1. Nome
6.2. lndirizzo
6.3. Numero di riconoscimento o di registrazione attribuito all'impresa o all'intermediario.
VI.7. Data.
Capitolo VII: Rinnovo dell'autorizzazione di additivi la cui autorizzazione è legata ad un responsabile dell'immissione in
circolazione
VII.1. Considerazioni generali.
Devono essere preparati un fascicolo ed una monografia aggiornati sulla base delle più aggiornate linee direttrici e deve essere fornito un elenco di tutte le variazioni di qualsiasi natura prodottesi successivamente alla concessione dell'autorizzazione all'immissione in circolazione o al suo ultimo rinnovo.
Deve essere confermato che la monografia ed il fascicolo sulla sicurezza siano stati adattati in modo da comprendere tutte le nuove informazioni concernenti l'additivo oppure quelle ora richieste a seguito delle modifiche previste dalle presenti linee direttrici.
Devono essere fornite informazioni anche sulla situazione dell'autorizzazione in tutto il mondo e sul volume delle vendite.
VII.2. Identità del principio attivo e dell'additivo.
Devono essere fornite prove che indichino che la composizione, la purezza o l'attività dell'additivo non siano state modificate o alterate rispetto a quelle dell'additivo già autorizzato. Devono essere segnalati eventuali cambiamenti del processo di produzione.
VII.3. Efficacia.
Devono essere presentate prove che dimostrino che l'additivo conserva l'asserita efficacia nelle condizioni di allevamento esistenti nell'Unione europea all'epoca della richiesta del rinnovo dell'autorizzazione. Tra tale prove devono figurare un resoconto delle esperienze generali connesse con l'utilizzo dell'additivo ed il controllo del rendimento.
VII.4. Microbiologia.
Particolare attenzione deve essere dedicata al possibile sviluppo della resistenza agli antimicrobici nel corso dell'utilizzo a lungo termine nelle normali condizioni d'uso. Le prove devono essere pertanto condotte nelle condizioni reali d'uso in aziende agricole dove da più lungo tempo venga ordinariamente utilizzato l'additivo. Come batteri di prova potrebbe essere utilizzata una serie di comuni batteri intestinali, tra i quali devono figurare gli organismi endogeni ed esogeni Grampositivi e Gram-negativi di interesse.
Se le prove dimostrano un cambiamento nel modello della resistenza rispetto ai dati di partenza, i batteri resistenti devono essere esaminati per valutare la resistenza crociata agli antibiotici utilizzati nel trattamento delle malattie infettive dell'uomo e degli animali. I più importanti sono gli antibiotici che appartengono allo stesso gruppo dell'additivo, ma nella sperimentazione devono essere compresi anche altri gruppi di antibiotici.
Devono essere segnalati i risultati di appropriati programmi di sorveglianza.
VII.5. Sicurezza.
Devono essere presentate le prove che dimostrino che, alla luce delle conoscenze attuali, l'additivo resta sicuro per le specie bersaglio, i consumatori, gli operatori e l'ambiente nelle condizioni approvate. Deve essere fornito un aggiornamento in materia di sicurezza che copra il periodo successivo all'autorizzazione all'immissione in circolazione o all'ultimo rinnovo della medesima, con informazioni sui seguenti punti:
segnalazioni degli effetti indesiderati compresi incidenti (effetti precedentemente sconosciuti, gravi effetti di qualsivoglia natura, accresciuta incidenza di effetti noti) che abbiano interessato animali bersaglio, operatori e l'ambiente. La relazione sull'effetto sfavorevole deve comprendere la natura dell'effetto, il numero dei soggetti/organismi colpiti, l'esito, le condizioni di utilizzo e una valutazione della causalità;
segnalazioni di interazioni e contaminazioni crociate precedentemente sconosciute;
se del caso dati relativi alla sorveglianza dei residui;
qualsiasi altra informazione concernente la sicurezza dell'additivo.
Devono essere chiaramente indicate le motivazioni per cui non vengano fornite ulteriori informazioni su uno qualsiasi di questi fattori.
Capitolo VII: Nuovo richiedente che si basa sulla prima autorizzazione di un additivo, la cui autorizzazione è legata ad un responsabile dell'immissione in circolazione.
Considerato che è possibile basarsi sulla valutazione dei dati forniti per una prima autorizzazione, un fascicolo predisposto per una richiesta ai sensi degli articoli 6 e 7, del decreto del Presidente della Repubblica n. 433, deve soddisfare unicamente i requisiti sottoindicati.
In questo senso un additivo può essere considerato identico se la composizione qualitativa e quantitativa nonché la purezza dei componenti attivi ed inattivi sono essenzialmente simili, se il preparato è lo stesso e le condizioni di impiego sono identiche.
Per tali prodotti non sarà di norma necessario ripetere gli studi farmacologici, tossicologici e d'efficacia e potrà essere presentata una richiesta in forma abbreviata, che deve comprendere le relazioni di esperti.
Devono essere presentate una relazione completa redatta secondo i criteri stabiliti nel cap. II di questo allegato ed una monografia.
Devono essere forniti dati analitici che indichino che l'additivo abbia delle specifiche chimicofisiche (parametri) simili a quelle del prodotto già autorizzato.
Deve essere confermato che le ulteriori conoscenze scientifiche, così come esse emergono dalla letteratura disponibile sull'additivo, non hanno modificato la precedente valutazione dell'efficacia dall'epoca della concessione dell'autorizzazione per l'immissione in circolazione dell'additivo.
Particolare attenzione deve essere dedicata al possibile sviluppo della resistenza agli antimicrobici nel corso dell'utilizzo a lungo termine del principio attivo nelle normali condizioni d'uso. Le prove devono essere pertanto condotte nelle condizioni reali d'uso in aziende agricole dove da più lungo tempo venga ordinariamente utilizzato l'additivo. Come batteri di prova potrebbe essere utilizzata una serie di comuni batteri intestinali, tra i quali devono figurare gli organismi endogeni ed esogeni Gram-positivi e Gram-negativi di interesse.
Se le prove dimostrano un cambiamento nel modello della resistenza rispetto ai dati di partenza, i batteri resistenti devono essere esaminati per valutare la resistenza crociata agli antibiotici utilizzati nel trattamento delle malattie infettive dell'uomo e degli animali. I più importanti sono gli antibiotici che appartengono allo stesso gruppo dell'additivo, ma nella sperimentazione devono essere compresi anche altri gruppi di antibiotici.
Devono essere presentate le prove che dimostrino che, alla luce delle conoscenze presenti, l'additivo resta sicuro per le specie bersaglio, i consumatori, gli operatori e l'ambiente nelle condizioni approvate. - Deve essere stabilito che il periodo di sospensione è conforme all'MRL.
[1] Abrogato dall'art. 18 del
[2] Numero così modificato da errata-corrige pubblicato nella G.U. 13 aprile 2002, n. 87.
[3] Numero così modificato da errata-corrige pubblicato nella G.U. 13 aprile 2002, n. 87.
[4] Numero così modificato da errata-corrige pubblicato nella G.U. 13 aprile 2002, n. 87.
[5] Numero così modificato da errata-corrige pubblicato nella G.U. 13 aprile 2002, n. 87.
[6] Numero così modificato da errata-corrige pubblicato nella G.U. 13 aprile 2002, n. 87.
[7] Numero così modificato da errata-corrige pubblicato nella G.U. 13 aprile 2002, n. 87.
[8] Numero così modificato da errata-corrige pubblicato nella G.U. 13 aprile 2002, n. 87.
[9] Allegato così sostituito dall'art. 1 del D.M. 19 luglio 2002.